I’m a Senior AI Research Scientist at Bevaya.ai (formerly Roots Automation). I train language and vision-language models at production scale, full-weight on an 8xH100 node, and the work centers on the data and evaluation side of it: benchmark construction, annotation design, calibration. Open-weights releases like GutenOCR ship with their training code and datasets. My roots are in scientific computing and molecular dynamics at Harvard, and that is where the work heads next: foundation-model training for the sciences. More about me →
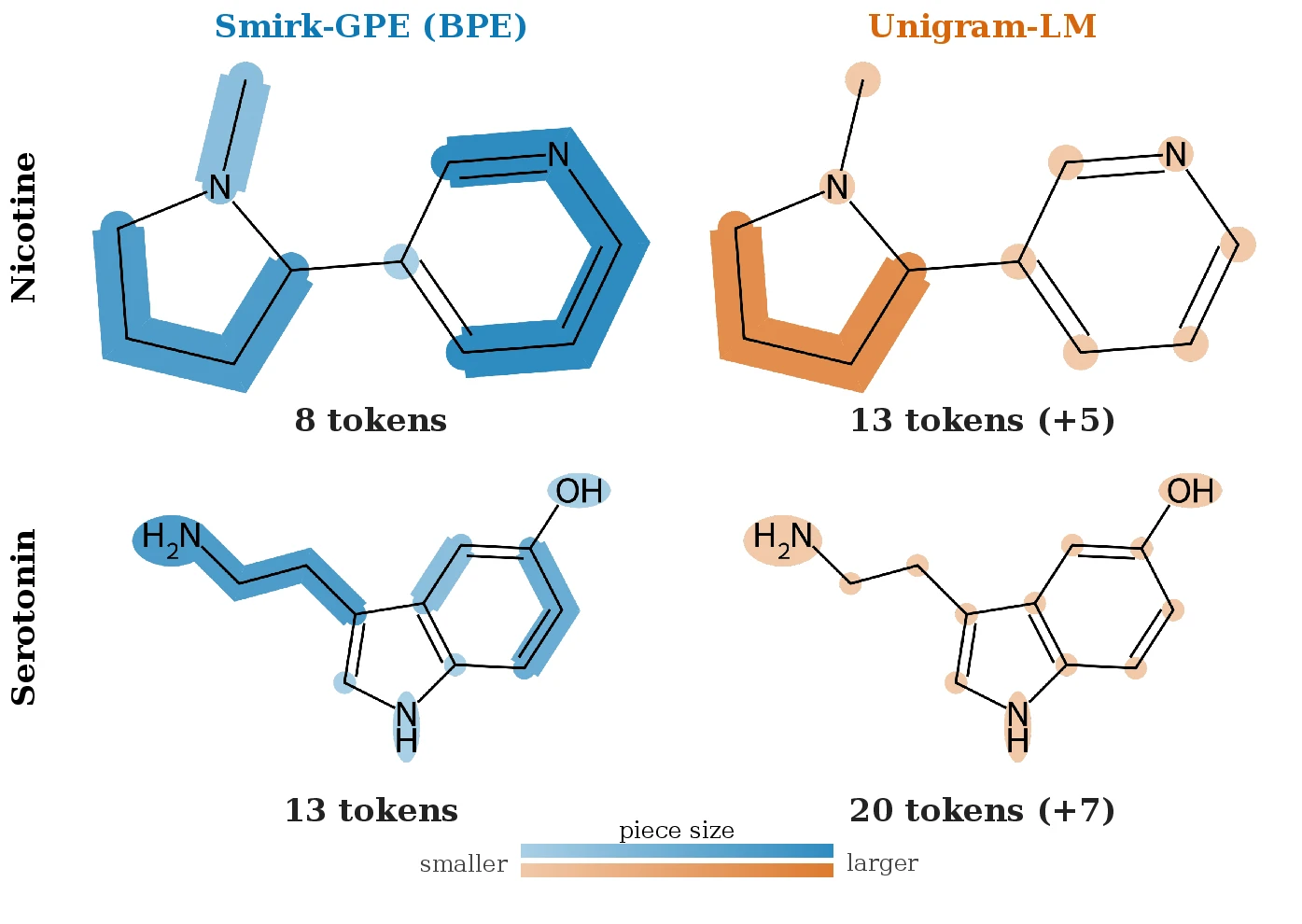
Where to Cut, How Deep: BPE and Unigram-LM on SMILES
A controlled comparison of BPE and Unigram-LM over a fixed chemistry SMILES glyph base. Across 22 matched conditions the two build near-disjoint subword vocabularies, so the subword algorithm is a modeling decision rather than a free default.

GutenOCR: A Grounded Vision-Language Front-End for Documents
GutenOCR is a family of vision-language models designed to serve as a ‘grounded OCR front-end’, providing high-quality text transcription and explicit geometric grounding.
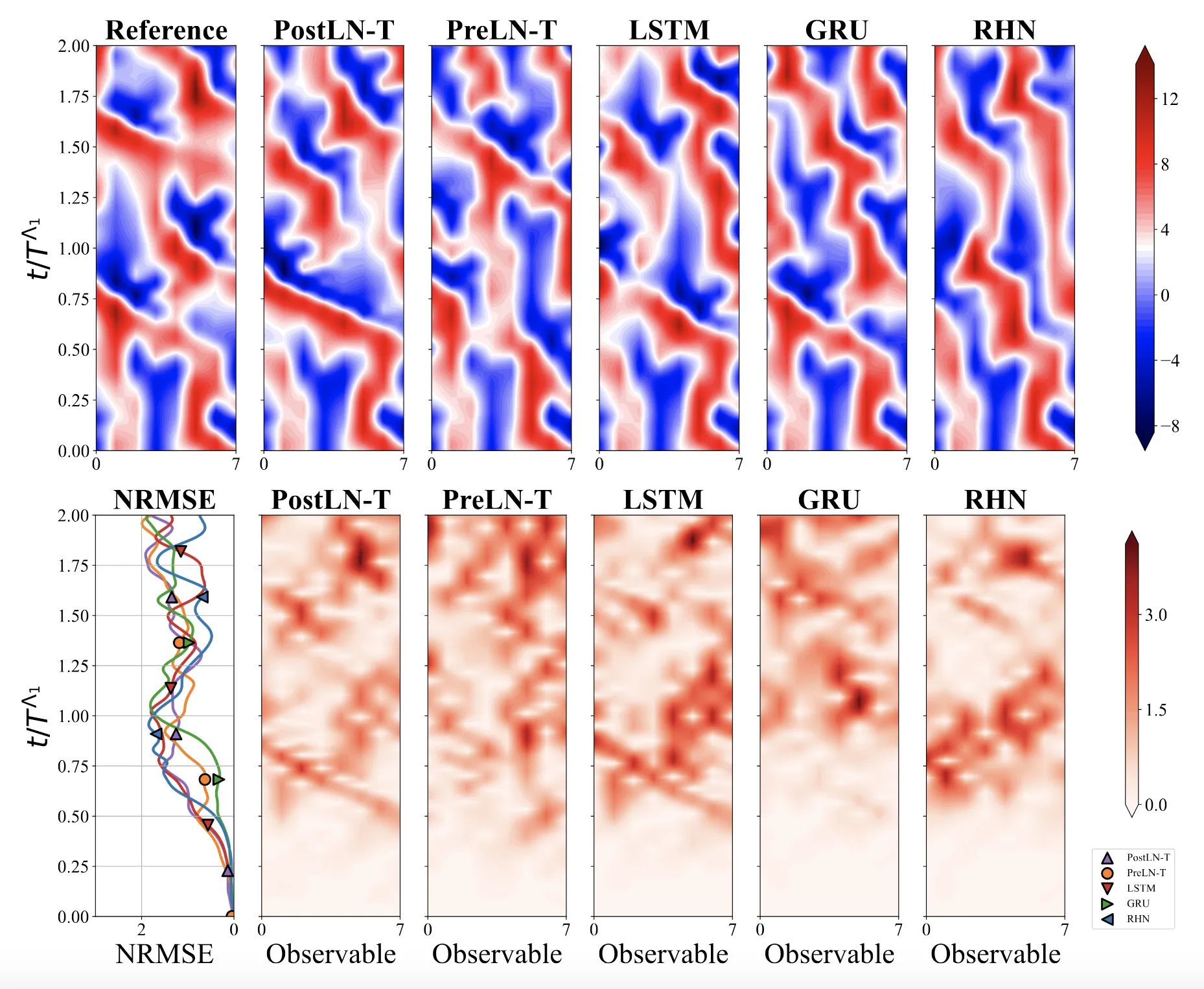
Optimizing Sequence Models for Dynamical Systems
We systematically ablate core mechanisms of Transformers and RNNs, finding that attention-augmented Recurrent Highway Networks outperform standard Transformers on forecasting high-dimensional chaotic systems.
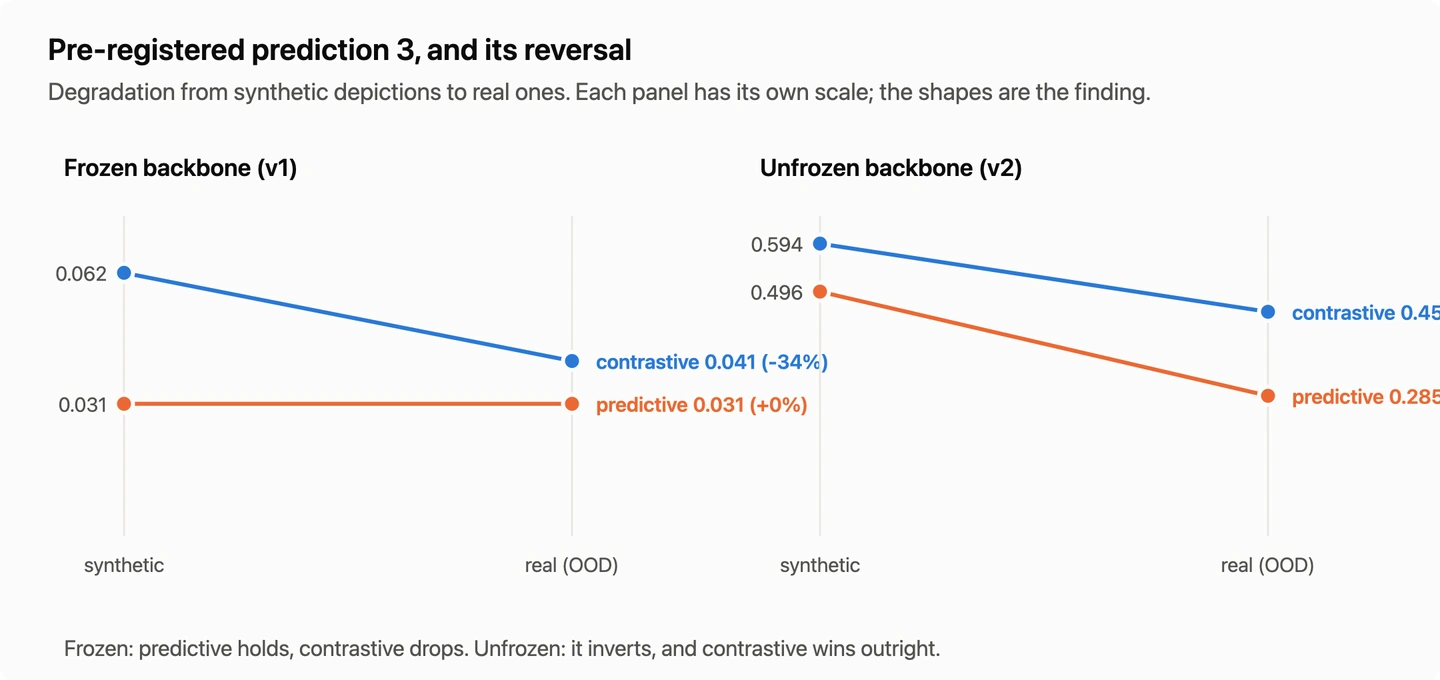
Molecular Depiction Alignment: Contrastive vs Predictive
Two ways of aligning molecular depictions into a frozen chemistry model’s embedding space, contrastive and predictive, compared on one data and evaluation stack with a pre-registered prediction.
What Surprised Me About Aligning Pictures of Molecules
I aligned a vision model into a frozen chemistry model’s embedding space two ways, contrastive and predictive, and held everything else identical. Four things surprised me, including a metric defect that produced exactly the result I had registered in advance.
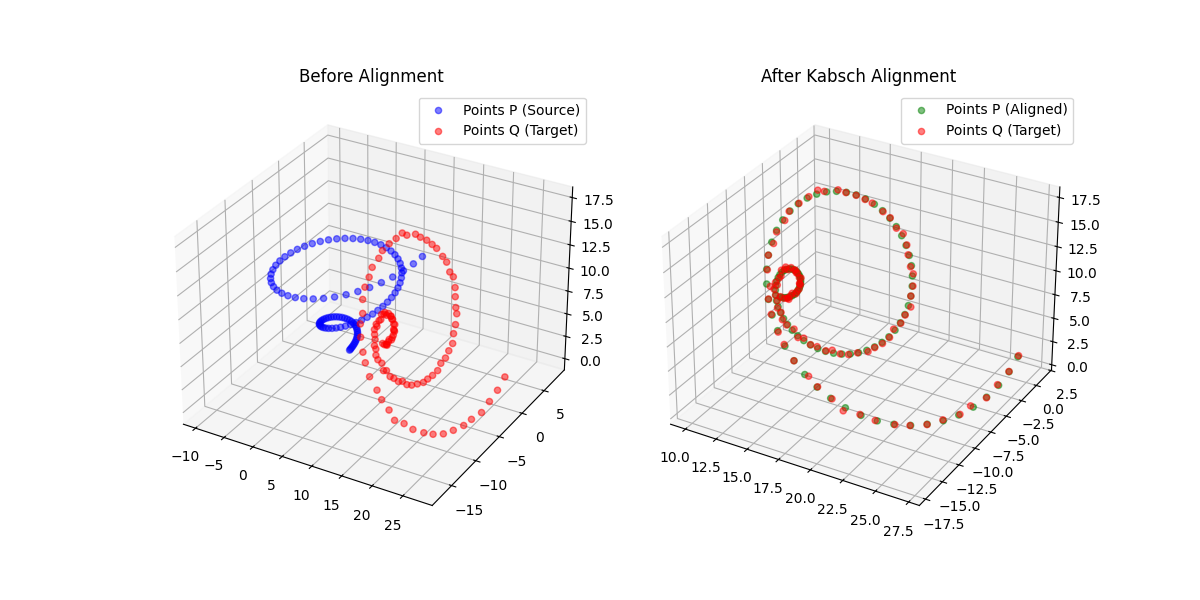
Kabsch-Horn Cookbook: Differentiable Alignment
A differentiable point-set alignment library implementing N-dimensional Kabsch, Horn quaternion, and Umeyama scaling algorithms with per-point weights, batch dimensions, and custom autograd across NumPy, PyTorch, JAX, TensorFlow, and MLX.
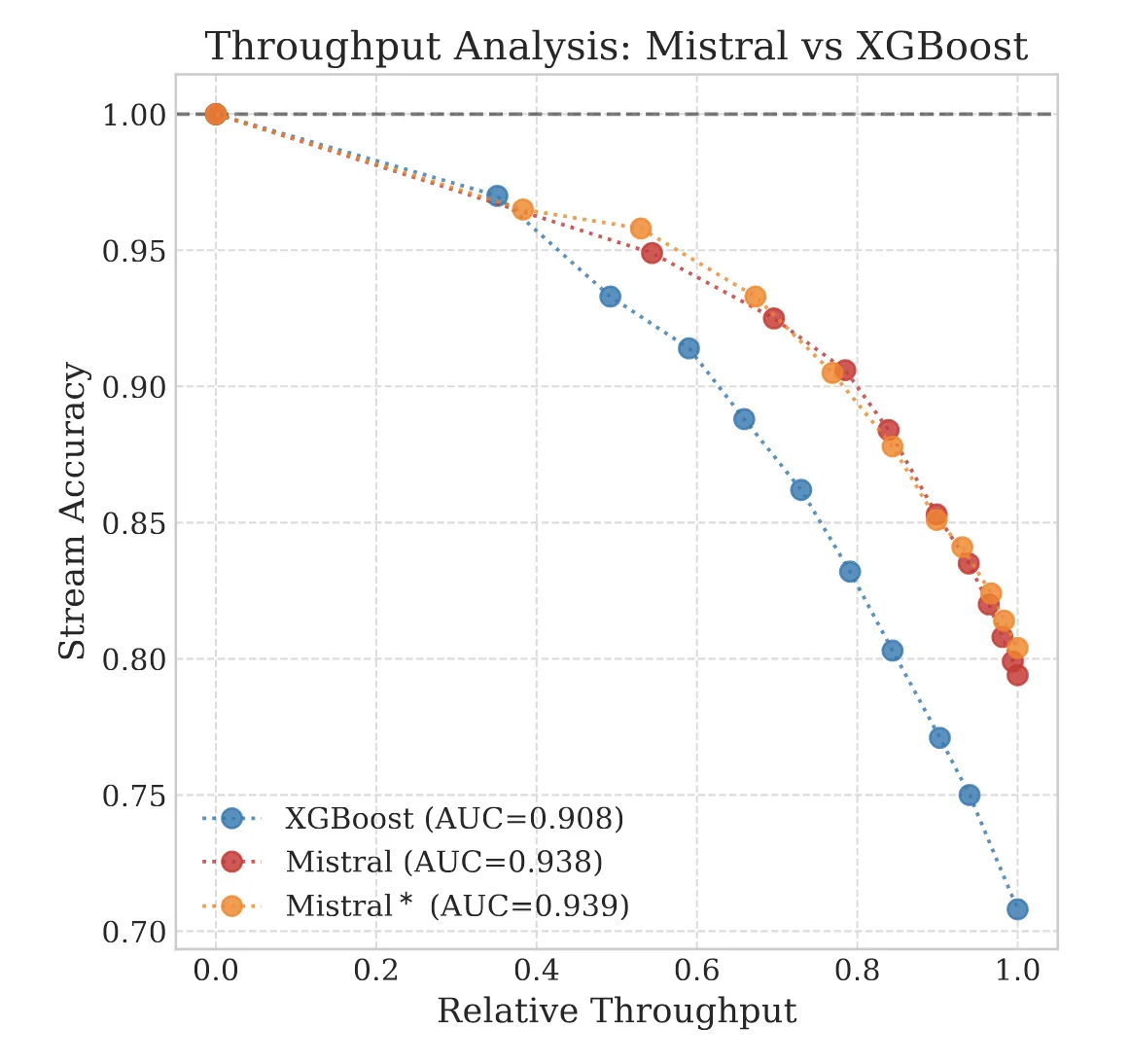
The Reliability Trap: The Limits of 99% Accuracy
We explore the ‘Silent Failure’ mode of LLMs in production: the limits of 99% accuracy for reliability, how confidence decays in long documents, and why standard calibration techniques struggle to fix it.
The Evolution of Page Stream Segmentation: Rules to LLMs
We trace the history of Page Stream Segmentation (PSS) through three eras (Heuristic, Encoder, and Decoder) and explain how privacy-preserving, localized LLMs enable true semantic processing.
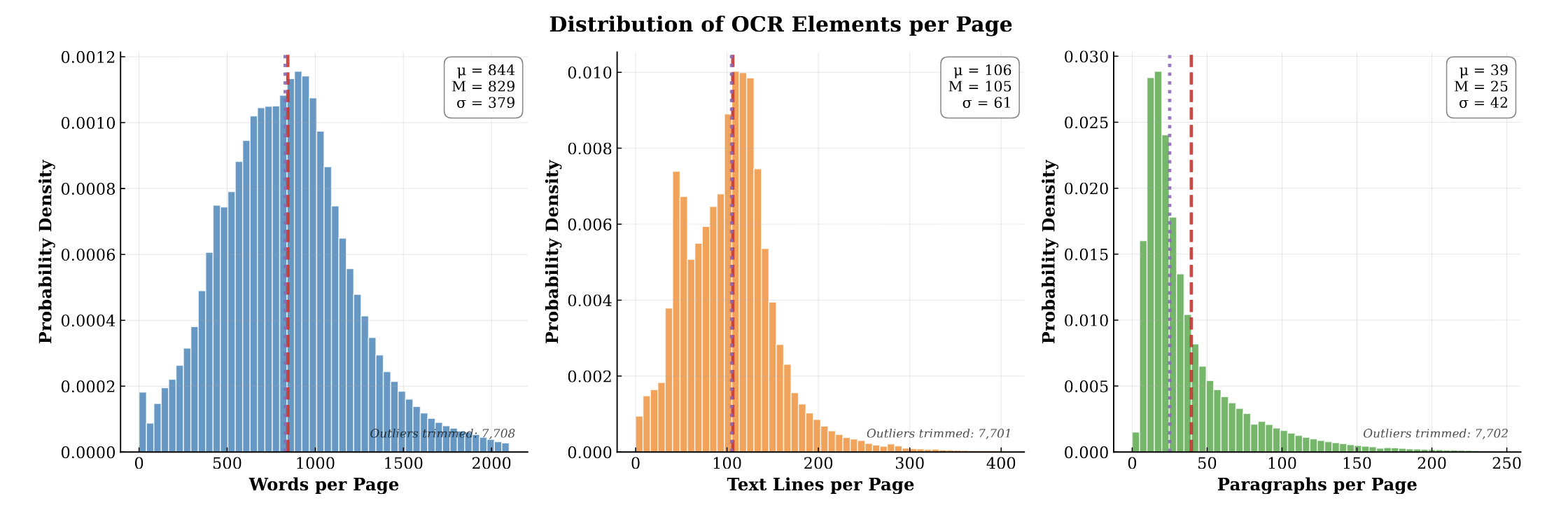
PubMed-OCR: PMC Open Access OCR Annotations
PubMed-OCR provides 1.5M pages of scientific articles with comprehensive OCR annotations and bounding boxes to support layout-aware modeling and document analysis.
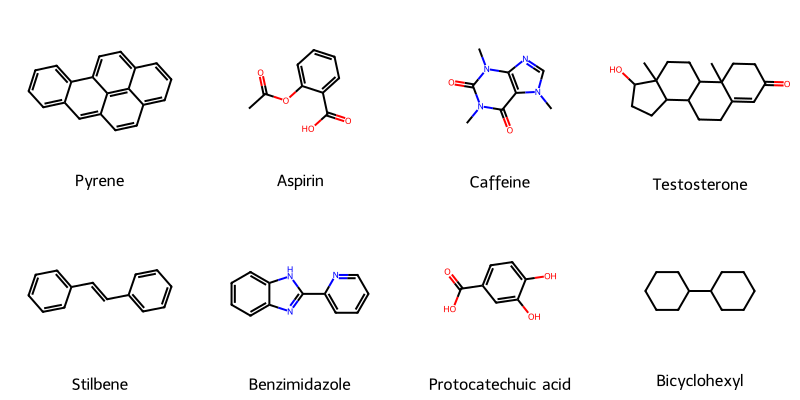
Molecular String Renderer: Chemical Visualization Library
An RDKit wrapper treating molecular visualization as a software engineering problem, implementing strategy pattern for SVG generation with automatic raster fallback, native SELFIES support for generative AI workflows, and strict type safety for batch processing in molecular ML training pipelines.

Importance Weighted Autoencoders: Beyond the Standard VAE
Discover how Importance Weighted Autoencoders (IWAEs) use the same architecture as VAEs with a different objective that optimizes a tighter bound on the log-likelihood, leveraging multiple samples effectively.
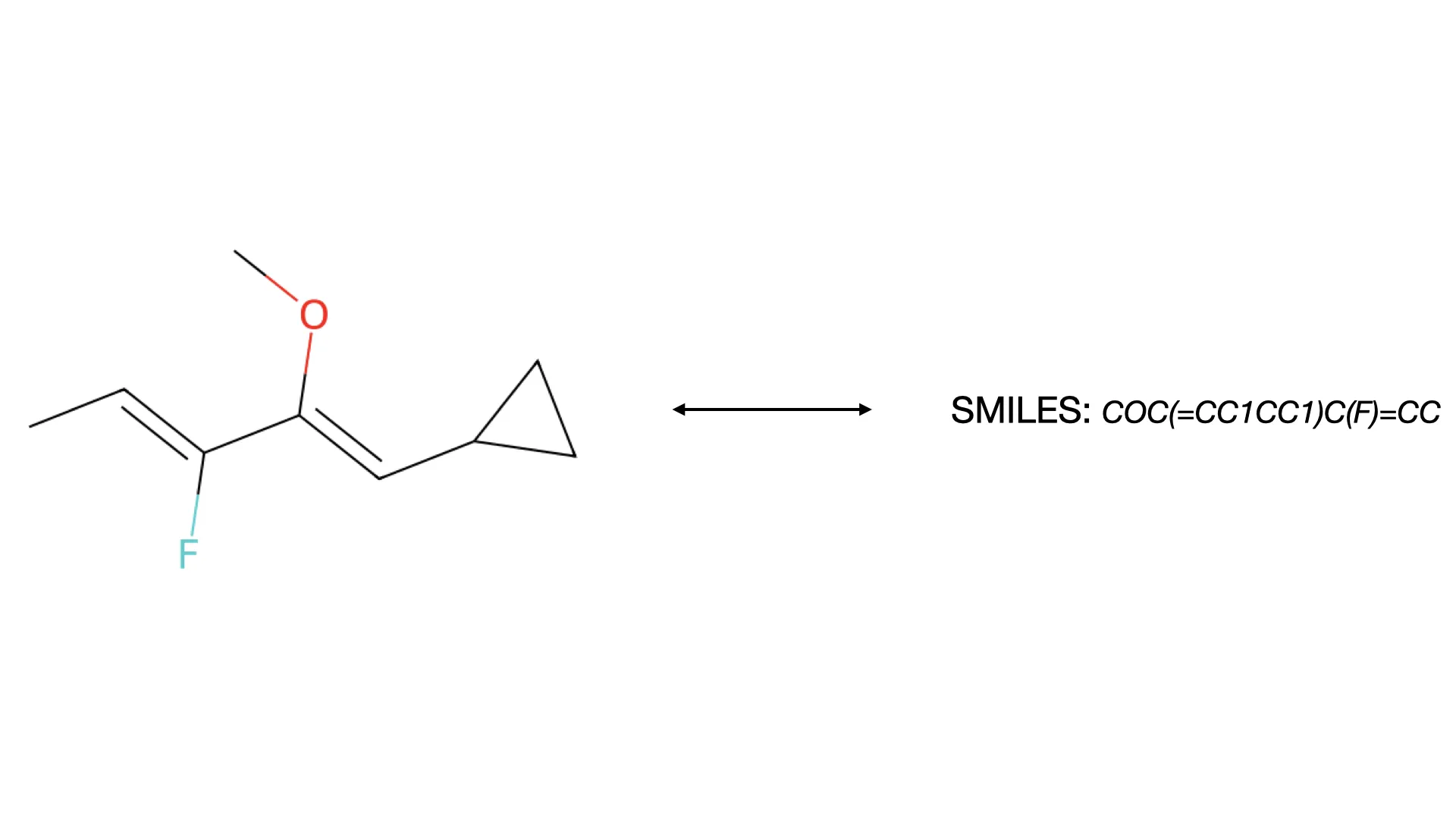
What is Optical Chemical Structure Recognition (OCSR)?
Discover how OCSR technology bridges the gap between molecular images and machine-readable data, evolving from rule-based systems to modern deep learning models for chemical knowledge extraction.