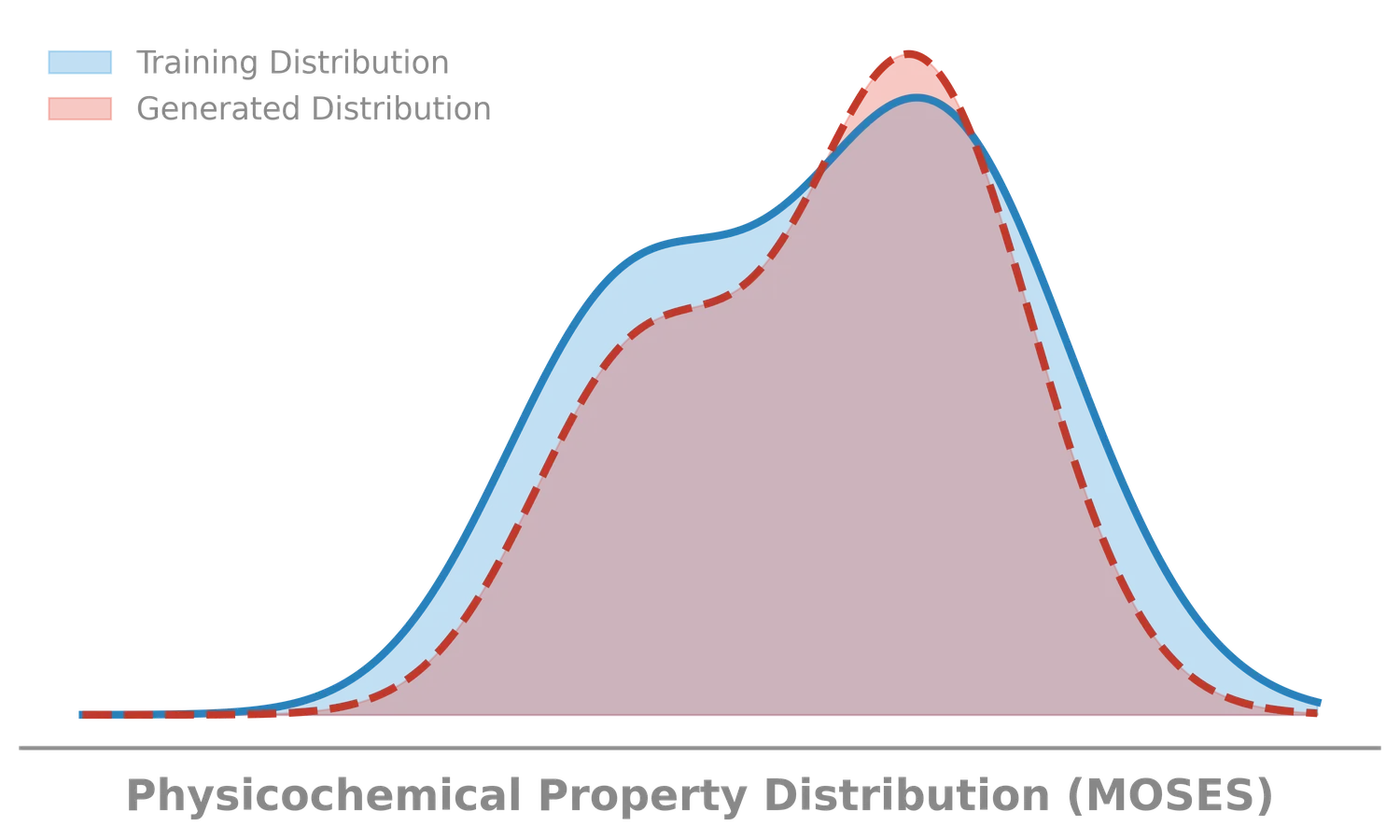
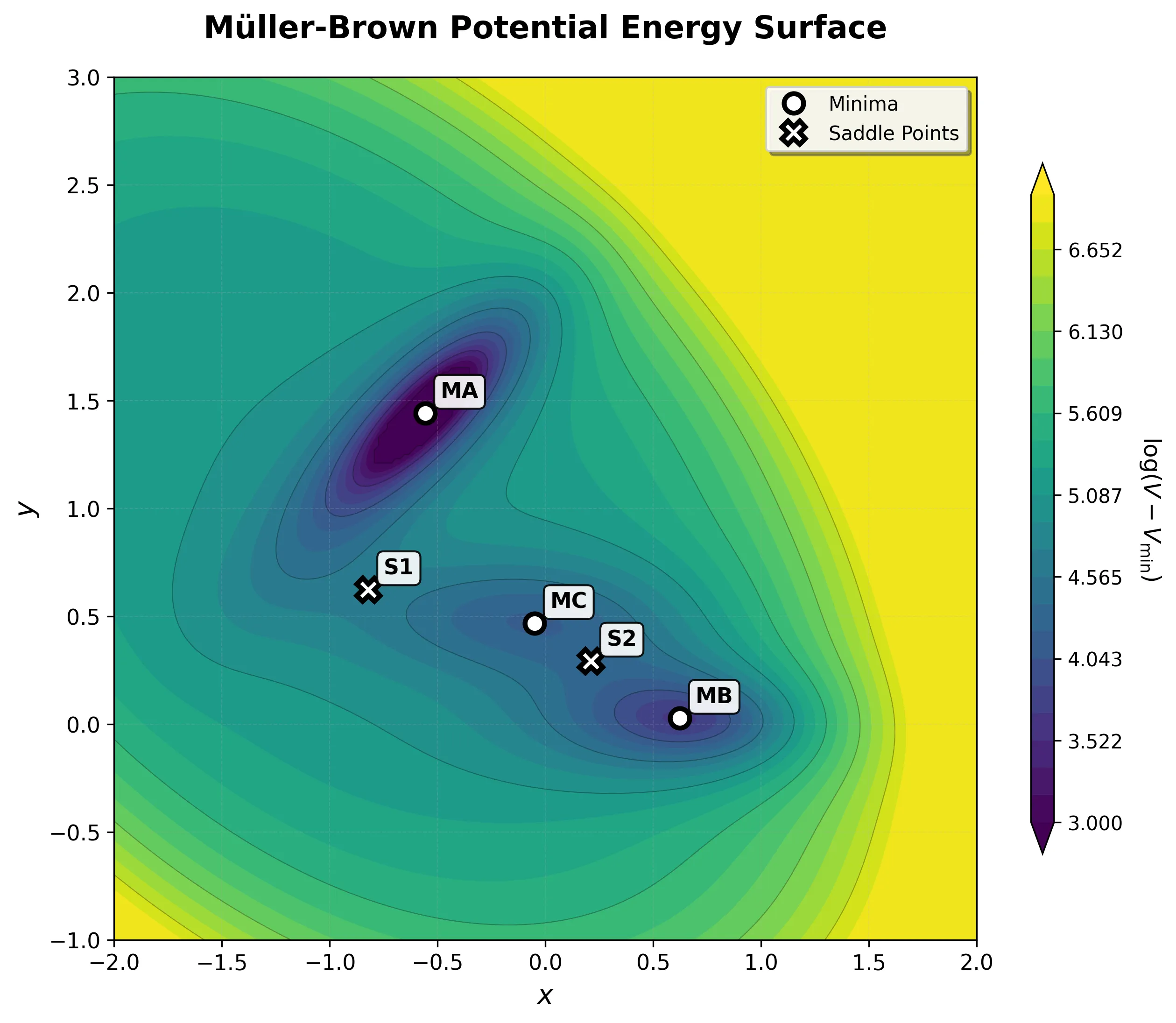
Progress in computational chemistry depends on shared, reproducible evaluation targets. This section collects notes on benchmark problems and datasets used to assess new methods, from classic analytical potential energy surfaces like the Muller-Brown surface to standardized generative modeling platforms like MOSES. These resources matter because they define what “better” means in practice, and understanding their design choices is essential for interpreting results reported in the literature.
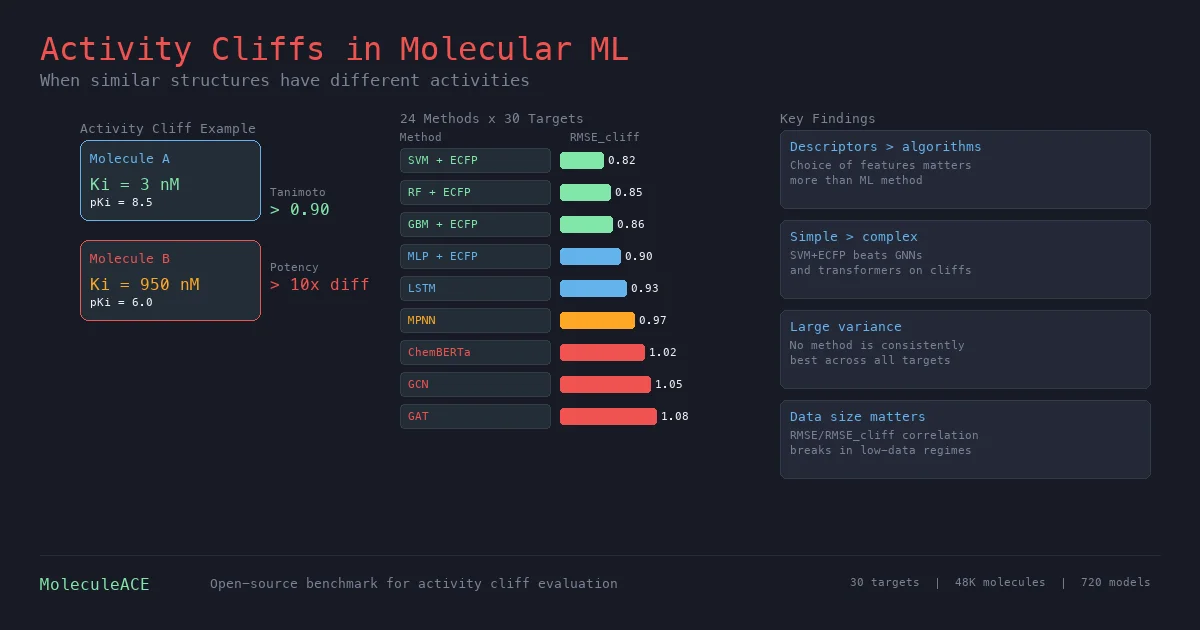
Exposing Limitations of Molecular ML with Activity Cliffs
This paper benchmarks 24 machine and deep learning methods on activity cliff compounds (structurally similar molecules with large potency differences) across 30 macromolecular targets. Traditional ML with molecular fingerprints consistently outperforms graph neural networks and SMILES-based transformers on these challenging cases, especially in low-data regimes.