What kind of paper is this?
This is technically a transcription of a conference talk, not a paper Levinthal wrote himself. The proceedings page credits “Notes by: A. Rawitch, Retranscribed: B. Krantz”, meaning what we have is a third-party record of an oral presentation Levinthal gave at the 1969 Mössbauer Spectroscopy in Biological Systems meeting at Allerton House, Illinois. This explains the informal, conversational register and the attached Q&A discussion.
In terms of contribution type, it functions as a Position paper (with Theory and Discovery elements):
- Position: Defines a “Grand Challenge” and argues for a conceptual shift in how we view biomolecular assembly
- Theory: Uses formal combinatorial arguments to establish the bounds of the search space ($10^{300}$ configurations)
- Discovery: Uses experimental data on alkaline phosphatase to validate the kinetic hypothesis
What is the motivation?
The Central Question: How does a protein choose one unique structure out of a hyper-astronomical number of possibilities in a biological timeframe (seconds)?
Levinthal provides a “back-of-the-envelope” derivation to define the problem scope:
- Degrees of Freedom: A generic, unrestricted protein with 2,000 atoms would possess ~6,000 degrees of freedom. However, physical constraints (specifically the planar peptide bond) reduce this significantly. For a 150-amino acid protein, these constraints lower the complexity to ~450 degrees of freedom (300 rotations, 150 bond angles).
- The Combinatorial Explosion: Even with conservative estimates, this results in $10^{300}$ possible conformations.
- The Time Constraint: Since proteins fold in seconds, Levinthal argues they can sample at most $10^8$ conformations (“postulating a minimum time from one conformation to another”) before stabilizing. Against $10^{300}$ possibilities, this search effectively covers 0% of the space, proving the impossibility of random search.
The Insight: The existence of folded proteins proves the impossibility of random global search. The system must be guided.
What is the novelty here?
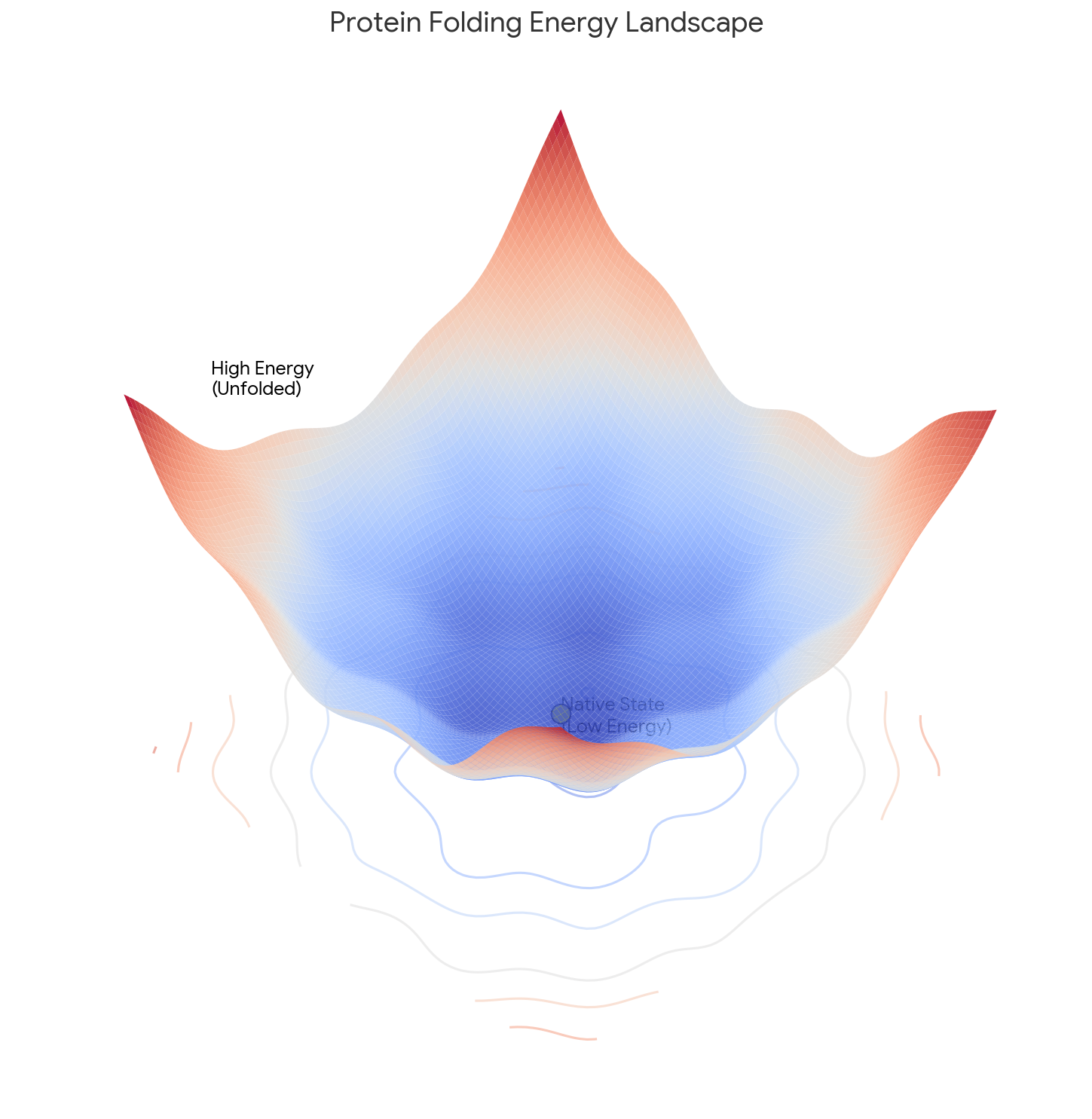
Core Contribution: Levinthal reframes folding from a thermodynamic problem (seeking the absolute global minimum) to a Kinetic Control problem. He argues the native state is a “metastable” energy well found quickly by a specific pathway, which can differ from the system’s lowest possible energy state.
The Pathway Dependence Hypothesis
The key insights of kinetic control:
- Nucleation: The process is “speeded and guided by the rapid formation of local interactions”
- Pathway Constraints: Local amino acid sequences form stable interactions and serve as nucleation points in the folding process, restricting the conformational search space
- The “Metastable” State: The final structure represents a “metastable state” in a sufficiently deep energy well that is kinetically accessible via the folding pathway, independent of the global energy minimum. Think of a ball that rolls into a valley on the side of a hill and stays there: it is not in the lowest valley on the entire landscape, but it is stable enough that it never escapes.

What experiments were performed?
To support the pathway hypothesis, Levinthal cites work on Alkaline Phosphatase (MW ~40,000), utilizing its property as a dimer of two identical subunits:
- Renaturation Window: The wild-type enzyme refolds optimally at 37°C. However, mutants were isolated that only produce active enzyme (and renature) at temperatures below 37°C.
- Stability vs. Formation: Crucially, once folded, both the wild-type and mutant enzymes are stable up to 90°C.
- The Rate-Limiting Step: Levinthal notes that the rate-limiting step for activity is the formation of the dimer from monomers. This proves that the order of assembly (kinetic pathway) dictates the final structure, distinct from the final structure’s thermodynamic stability.
The talk concluded with a short motion picture Levinthal showed live, illustrating polypeptide synthesis and “the process of then forming a desired interaction via the most favored energy path as displayed on the computer controlled oscilloscope.”
The Q&A discussion following the talk includes one exchange directly relevant to the folding argument: when asked whether a protein is ever truly unfolded (devoid of all secondary and tertiary structure), Levinthal answered that both physical measurements and synthetic polypeptide work suggest yes. The other exchanges concerned the tangent formula for x-ray crystallographic phase refinement and whether computed structures had been tested for thermal perturbations.
What outcomes/conclusions?
Key Finding
The mutant experiments serve as the “smoking gun”: a protein seeking a global thermodynamic minimum would fold spontaneously at any temperature where the final state is stable (up to 90°C). The fact that mutants require specific lower temperatures for formation (while remaining stable at high temperatures once formed) proves that the kinetic pathway determines the outcome alongside the thermodynamic endpoint.
Broader Implications
Levinthal explicitly asks: “Is a unique folding necessary for any random 150-amino acid sequence?” and answers “Probably not.” He supports this by noting the difficulty many researchers face in attempting to crystallize proteins, suggesting that not all sequences produce stably folded structures.
He concludes by connecting these computational models to Mössbauer spectroscopy, suggesting that these computational studies may help in understanding how small perturbations of polypeptide structures affect the Mössbauer nucleus (a reminder of the specific conference context where this perspective was delivered).
Connection to Modern Work
Levinthal’s arguments remain relevant context for modern computational protein folding:
- Early computational visualization: Levinthal used computer-controlled oscilloscopes and vector matrix multiplications to build and display 3D polypeptide structures, and showed a motion picture of forming a desired interaction via the most favored energy path. This was an early instance of computational molecular visualization.
- Local interactions and folding pathways: The hypothesis that “local interactions” serve as nucleation points that guide folding remains central to how modern structure prediction methods (e.g., AlphaFold) model residue-residue interactions.
- The paradox’s lasting influence: The impossibility of random conformational search that Levinthal articulated continues to motivate approaches that exploit the structure of the energy landscape rather than exhaustive enumeration.
- Sequence-structure relationship: Levinthal’s suggestion that not every random amino acid sequence would fold uniquely foreshadows the modern challenge of inverse folding (protein design), where the goal is to find sequences within the subset that does fold to a target structure.
Paper Information
Citation: Levinthal, C. (1969). How to Fold Graciously. In Mössbauer Spectroscopy in Biological Systems: Proceedings of a meeting held at Allerton House, Monticello, Illinois (pp. 22-24). University of Illinois Press.
Publication: Mössbauer Spectroscopy in Biological Systems Proceedings, 1969
@inproceedings{levinthal1969fold,
title={How to fold graciously},
author={Levinthal, Cyrus},
booktitle={M{\"o}ssbauer spectroscopy in biological systems},
pages={22--24},
year={1969},
publisher={University of Illinois Press},
url={https://faculty.cc.gatech.edu/~turk/bio_sim/articles/proteins_levinthal_1969.pdf}
}
Additional Resources: