MolGen: Molecular Generation with Chemical Feedback
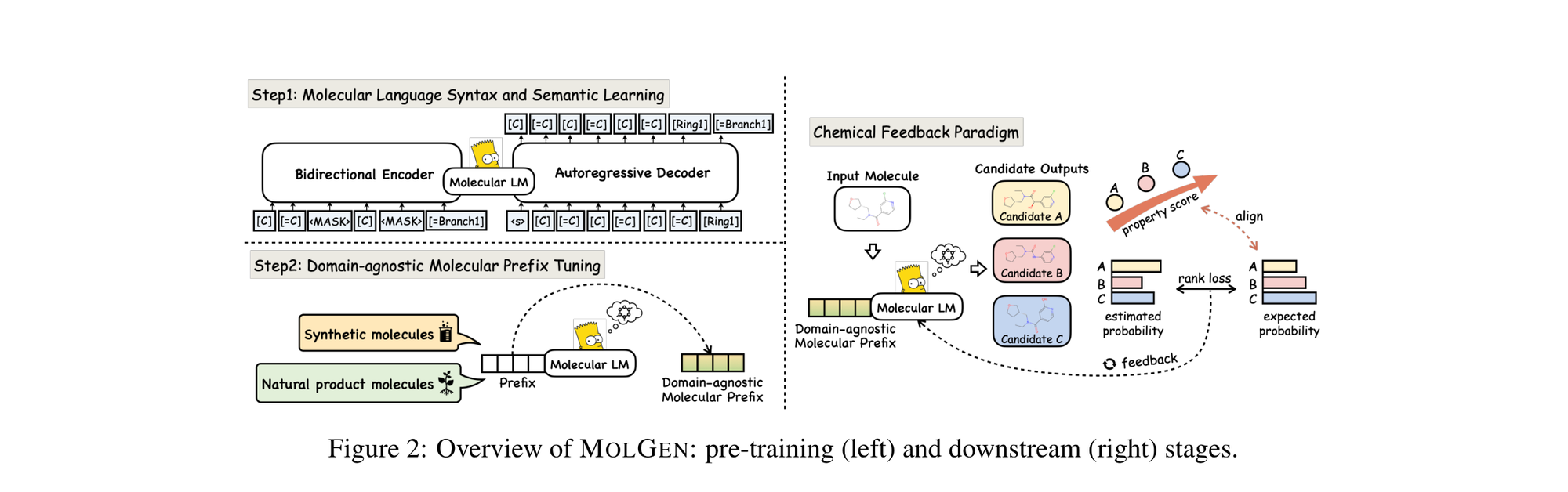
MolGen pre-trains on 100M+ SELFIES molecules, introduces domain-agnostic prefix tuning for cross-domain transfer, and applies a chemical feedback paradigm to reduce molecular hallucinations.
MolGen pre-trains on 100M+ SELFIES molecules, introduces domain-agnostic prefix tuning for cross-domain transfer, and applies a chemical feedback paradigm to reduce molecular hallucinations.
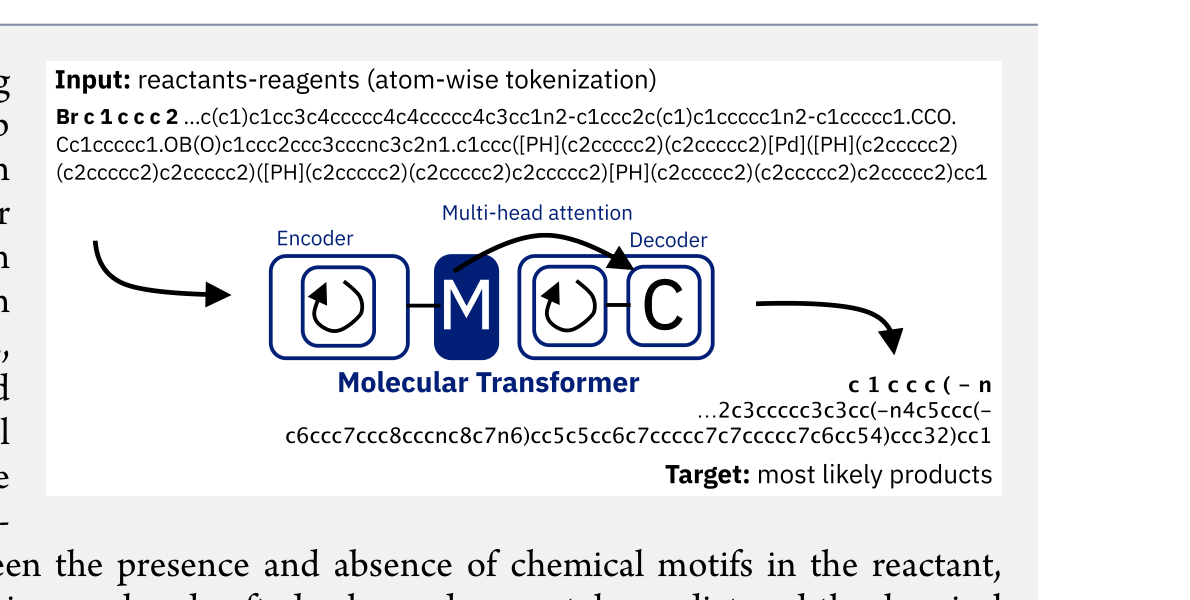
The Molecular Transformer applies the Transformer architecture to forward reaction prediction, treating it as SMILES-to-SMILES machine translation. It achieves 90.4% top-1 accuracy on USPTO_MIT, outperforms quantum-chemistry baselines on regioselectivity, and provides calibrated uncertainty scores (0.89 AUC-ROC) for ranking synthesis pathways.
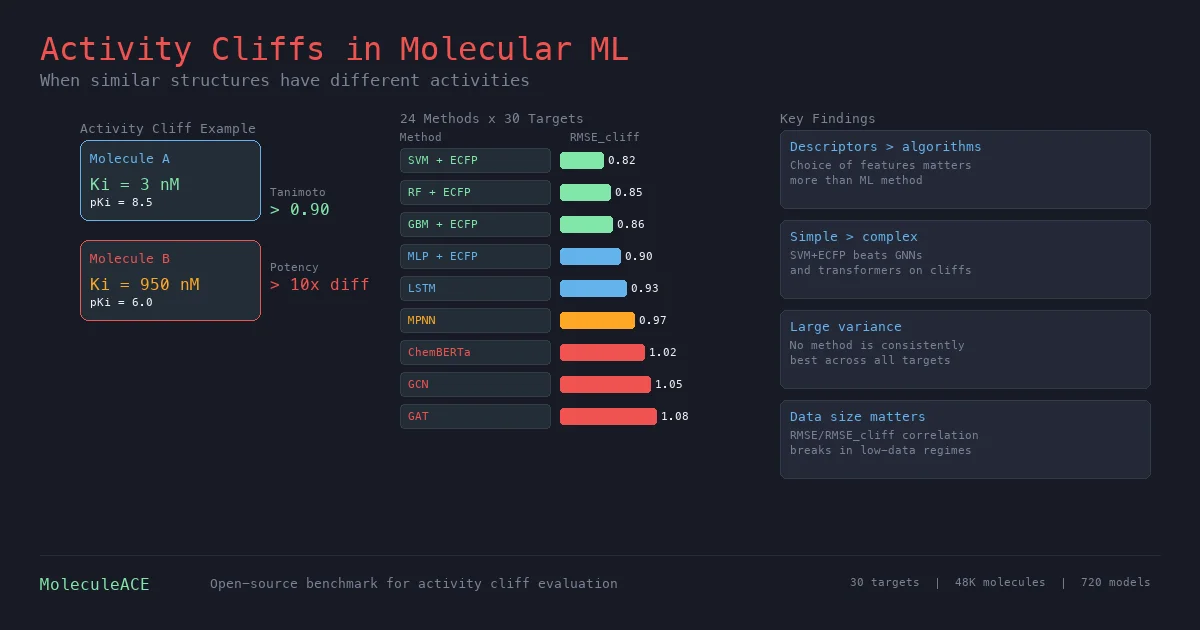
This paper benchmarks 24 machine and deep learning methods on activity cliff compounds (structurally similar molecules with large potency differences) across 30 macromolecular targets. Traditional ML with molecular fingerprints consistently outperforms graph neural networks and SMILES-based transformers on these challenging cases, especially in low-data regimes.
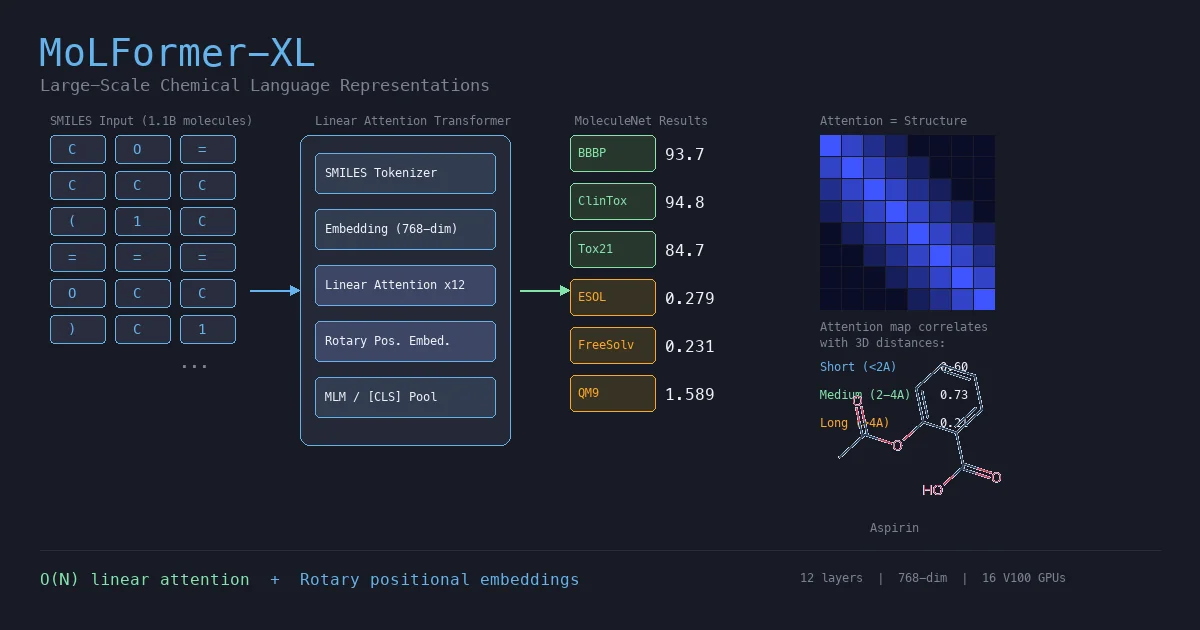
MoLFormer is a transformer encoder with linear attention and rotary positional embeddings, pretrained via masked language modeling on 1.1 billion molecules from PubChem and ZINC. MoLFormer-XL outperforms GNN baselines on most MoleculeNet classification and regression tasks, and attention analysis reveals that the model learns interatomic spatial relationships directly from SMILES strings.
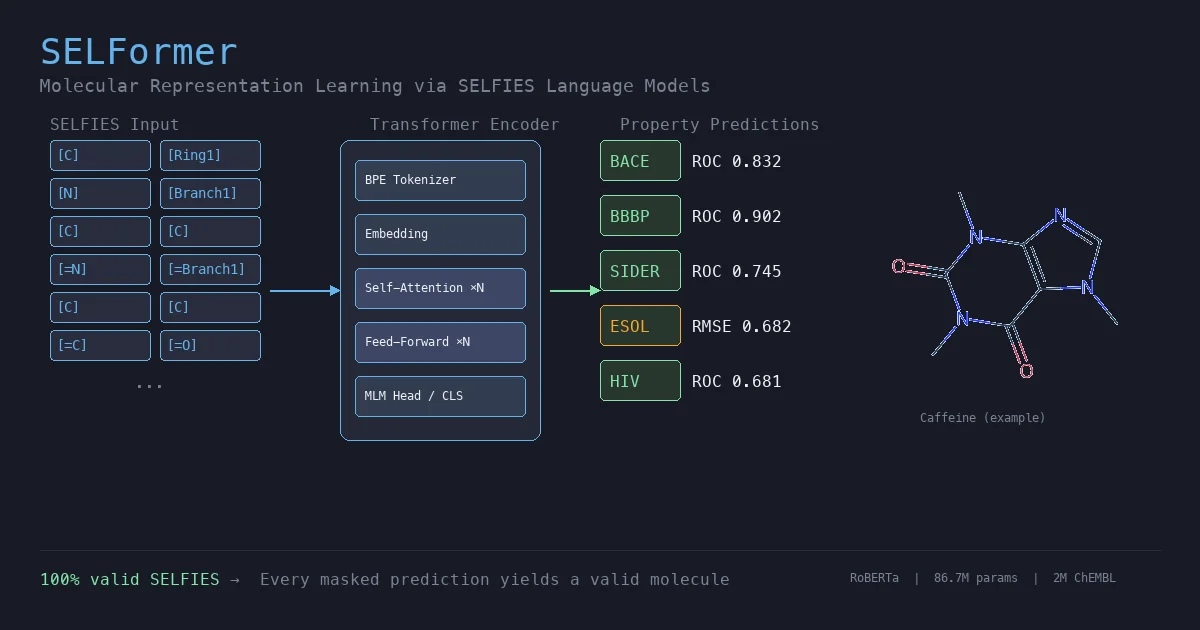
SELFormer is a transformer-based chemical language model that uses SELFIES instead of SMILES as input. Pretrained on 2M ChEMBL compounds via masked language modeling, it achieves strong classification performance on MoleculeNet tasks, outperforming ChemBERTa-2 by ~12% on average across BACE, BBBP, and HIV.
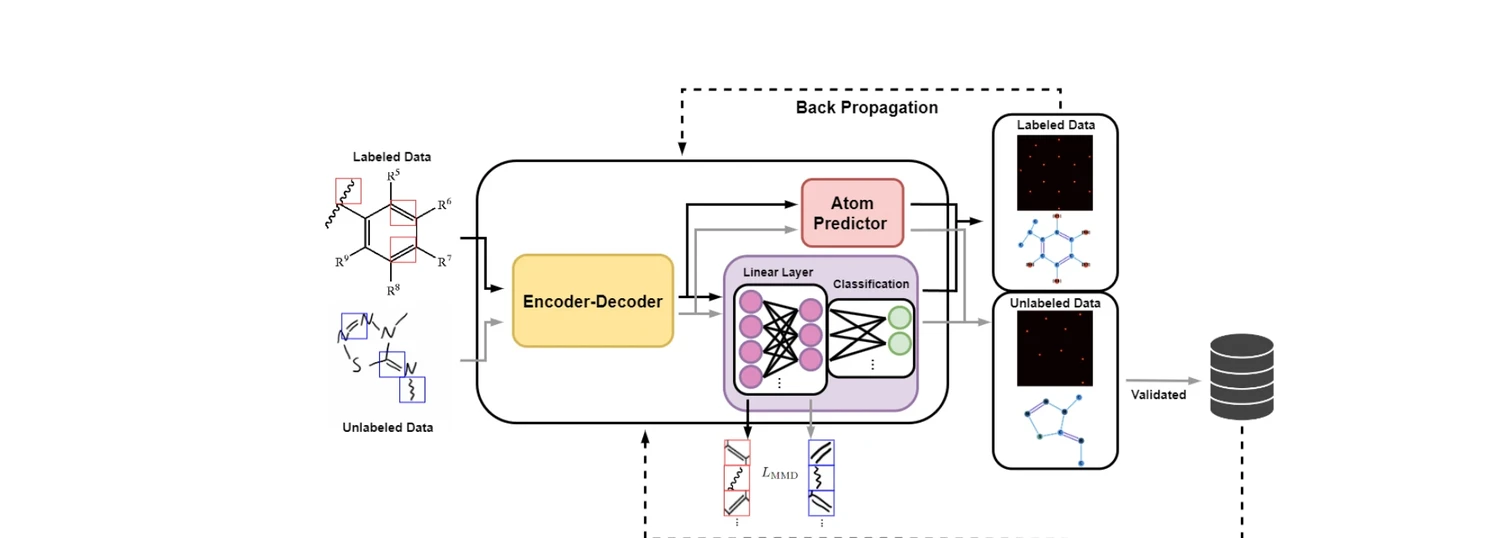
AdaptMol combines an end-to-end graph reconstruction model with unsupervised domain adaptation via class-conditional MMD on bond features and SMILES-validated self-training. Achieves 82.6% accuracy on hand-drawn molecules (10.7 points above prior best) while maintaining state-of-the-art results on four literature benchmarks, using only 4,080 real hand-drawn images for adaptation.
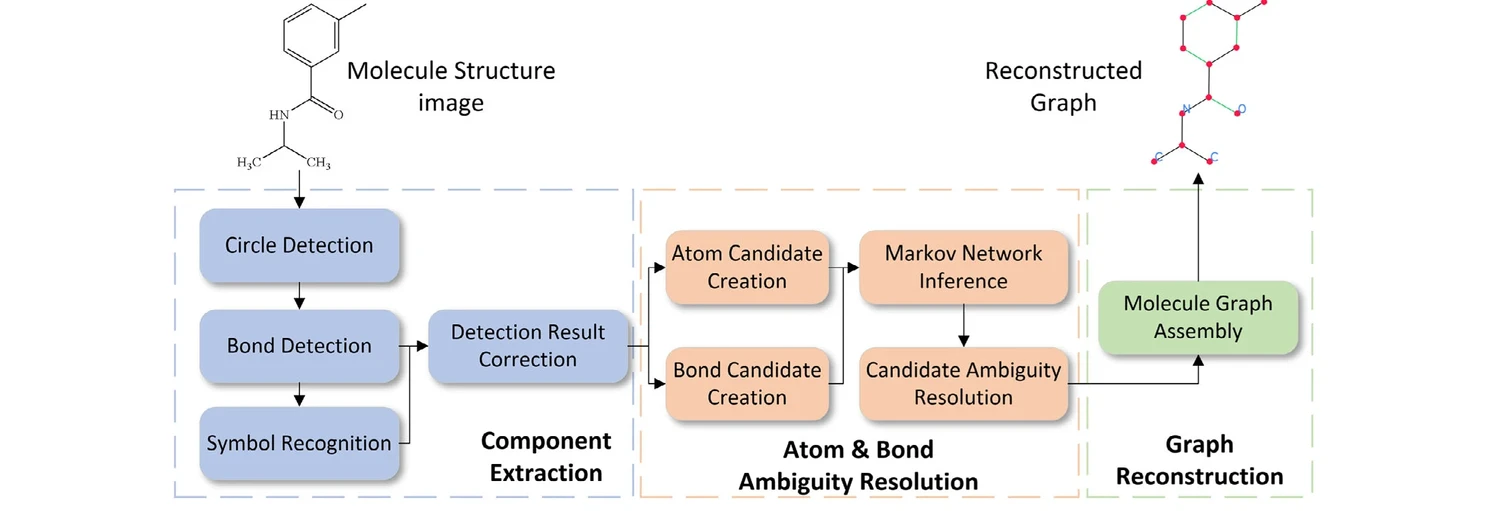
GraphReco presents a rule-based OCSR system with two key innovations: a Fragment Merging line detection algorithm for precise bond identification and a Markov network for probabilistic resolution of atom/bond ambiguity during graph assembly. Achieves 94.2% accuracy on USPTO-10K, outperforming both traditional rule-based and some ML-based methods.
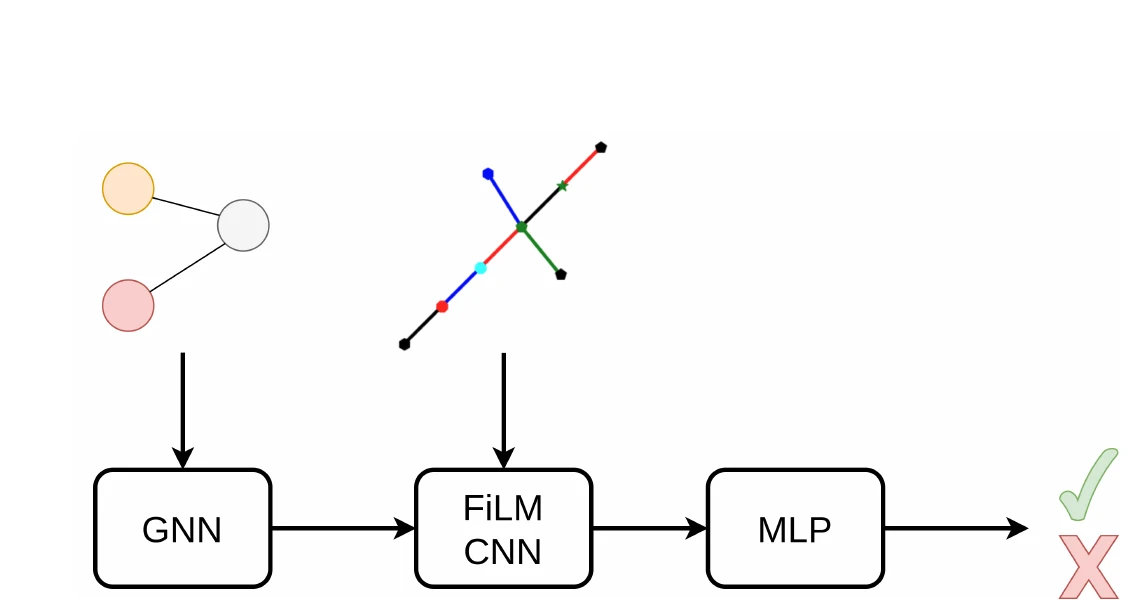
GraSP introduces a general framework for recognizing graphs in images by framing it as sequential subgraph prediction with a binary classifier. A GNN conditions a CNN via FiLM layers to predict whether a candidate graph is a subgraph of the target. Applied to OCSR on QM9, GraSP achieves 67.5% accuracy with no domain-specific modifications.
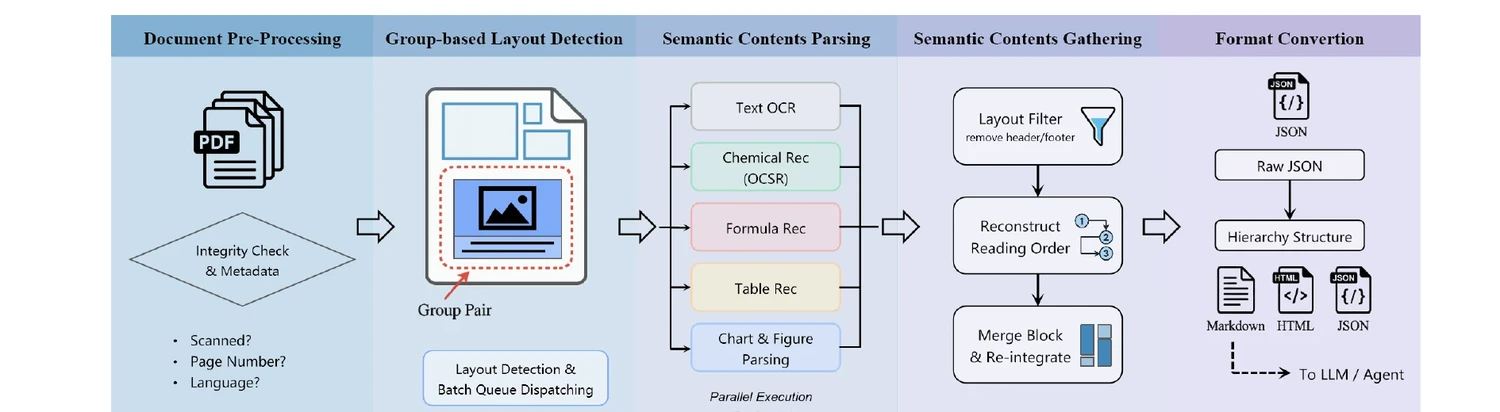
Technical report on Uni-Parser, an industrial-grade document parsing engine that uses a modular multi-expert architecture to parse scientific PDFs into structured representations. Integrates MolParser 1.5 for OCSR, achieving 88.6% accuracy on chemical structures while processing up to 20 pages per second.

ChemBERTa-3 provides a unified, scalable infrastructure for pretraining and benchmarking chemical foundation models. It addresses reproducibility gaps in previous studies like MoLFormer through standardized scaffold splitting and open-source tooling.
ChemDFM-R is a 14B-parameter chemical reasoning model that integrates a 101B-token dataset of atomized chemical knowledge. Using a mix-sourced distillation strategy and domain-specific reinforcement learning, it outperforms similarly sized models and DeepSeek-R1 on ChemEval.

This work investigates the scaling hypothesis for molecular transformers, training RoBERTa models on 77M SMILES from PubChem. It compares Masked Language Modeling (MLM) against Multi-Task Regression (MTR) pretraining, finding that MTR yields better downstream performance but is computationally heavier.