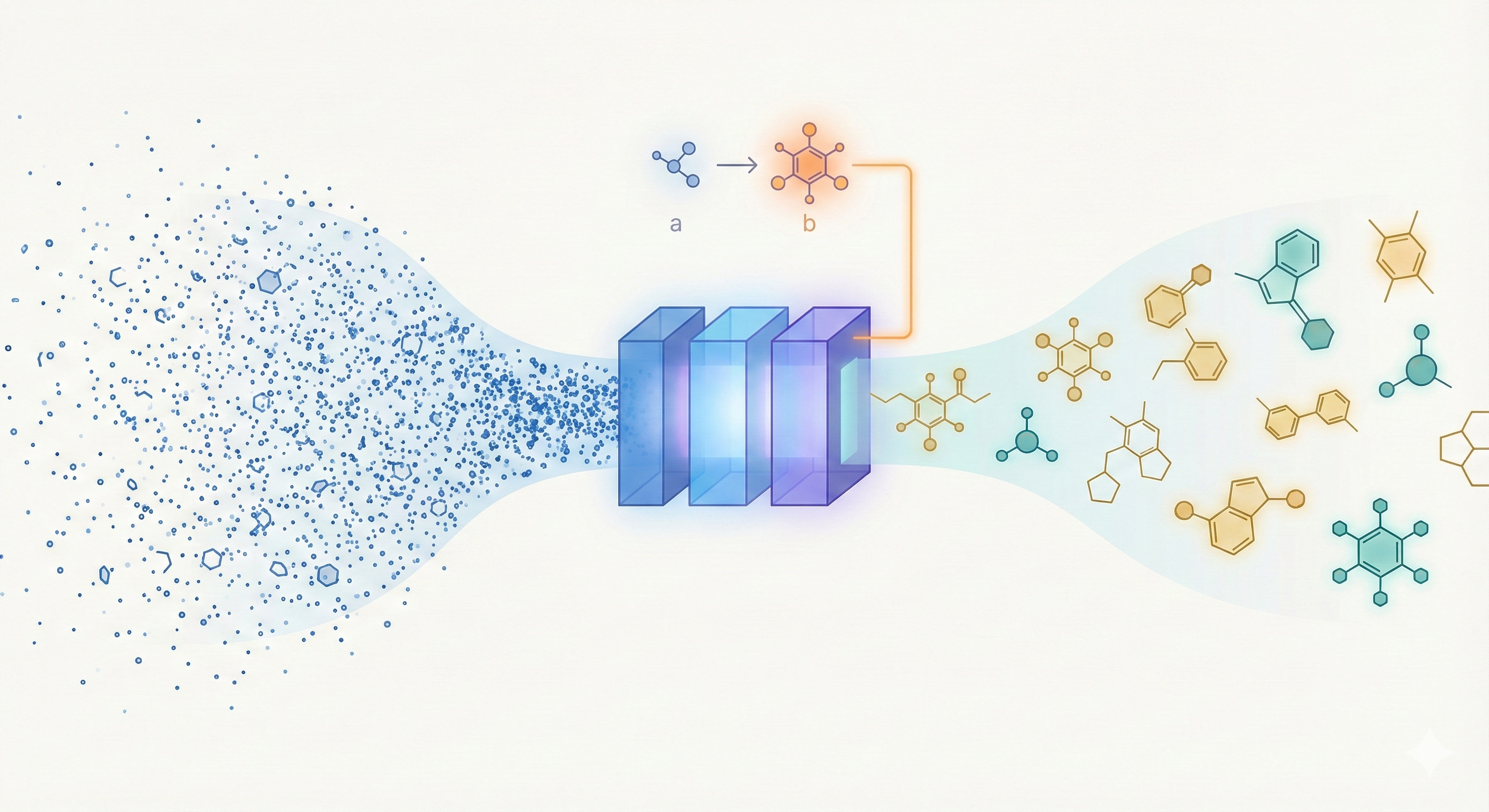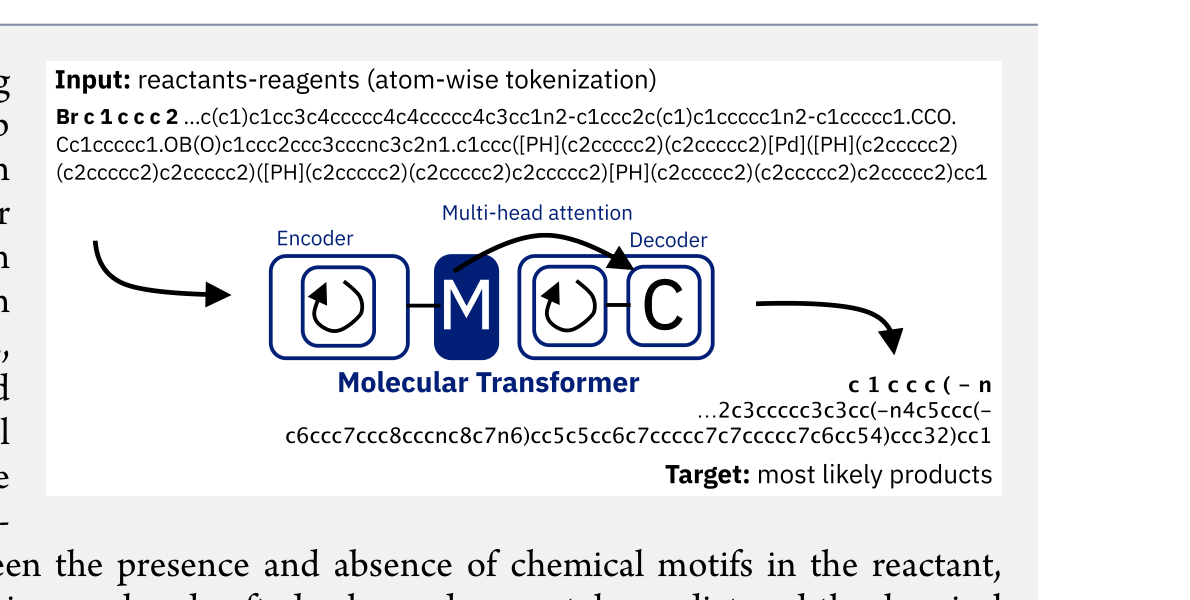
Molecular Transformer: Calibrated Reaction Prediction
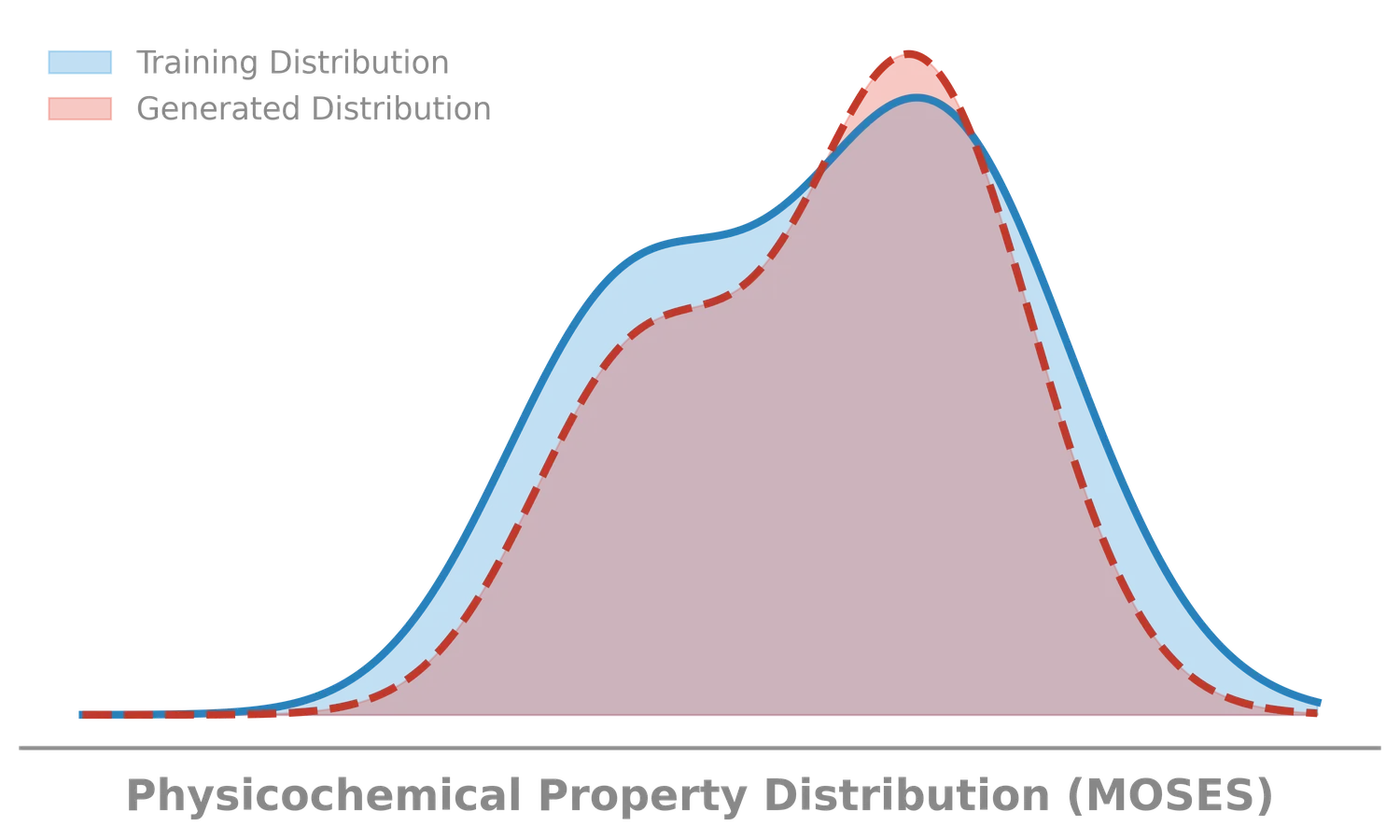
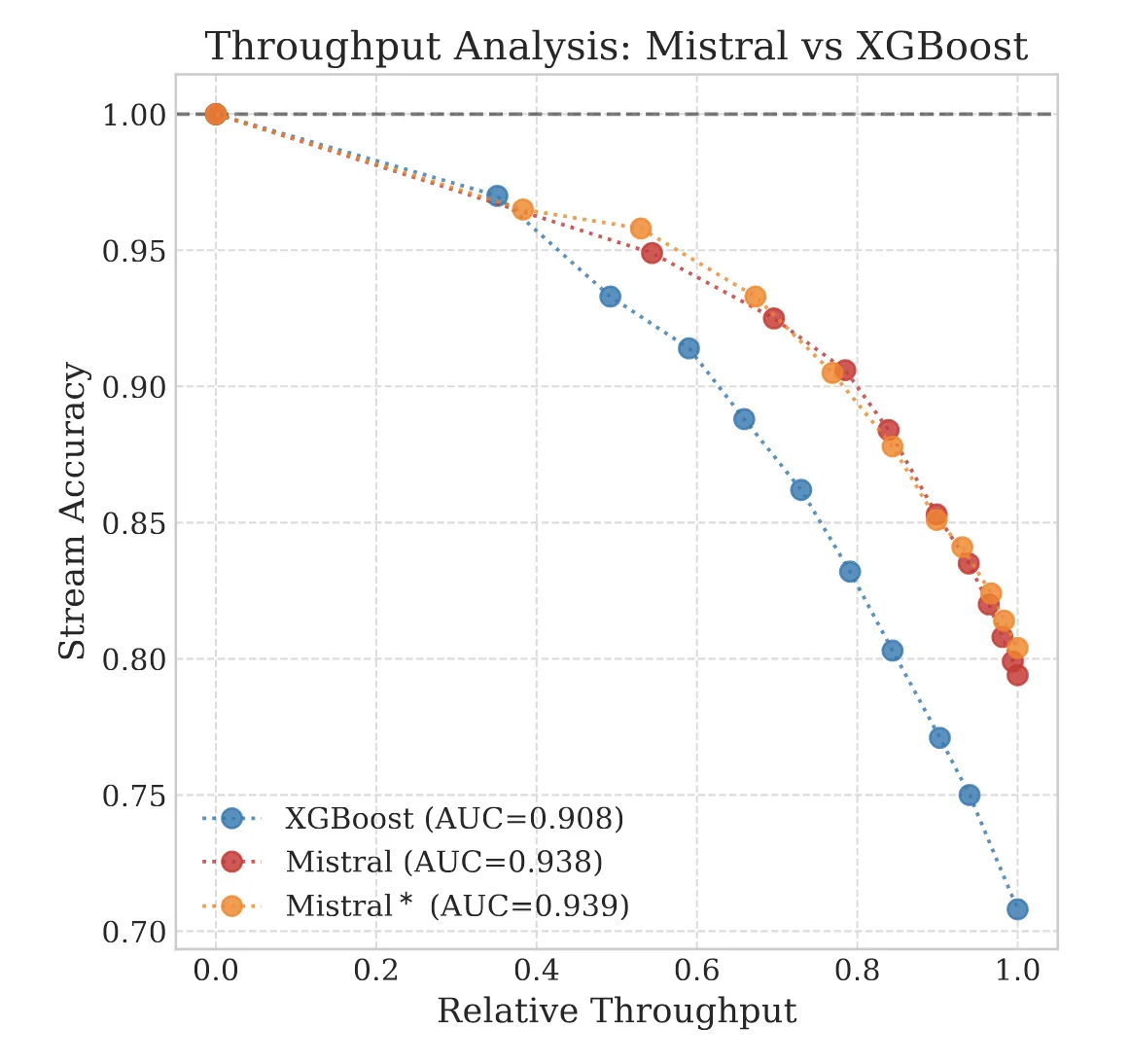
The Molecular Transformer applies the Transformer architecture to forward reaction prediction, treating it as SMILES-to-SMILES machine translation. It achieves 90.4% top-1 accuracy on USPTO_MIT, outperforms quantum-chemistry baselines on regioselectivity, and provides calibrated uncertainty scores (0.89 AUC-ROC) for ranking synthesis pathways.