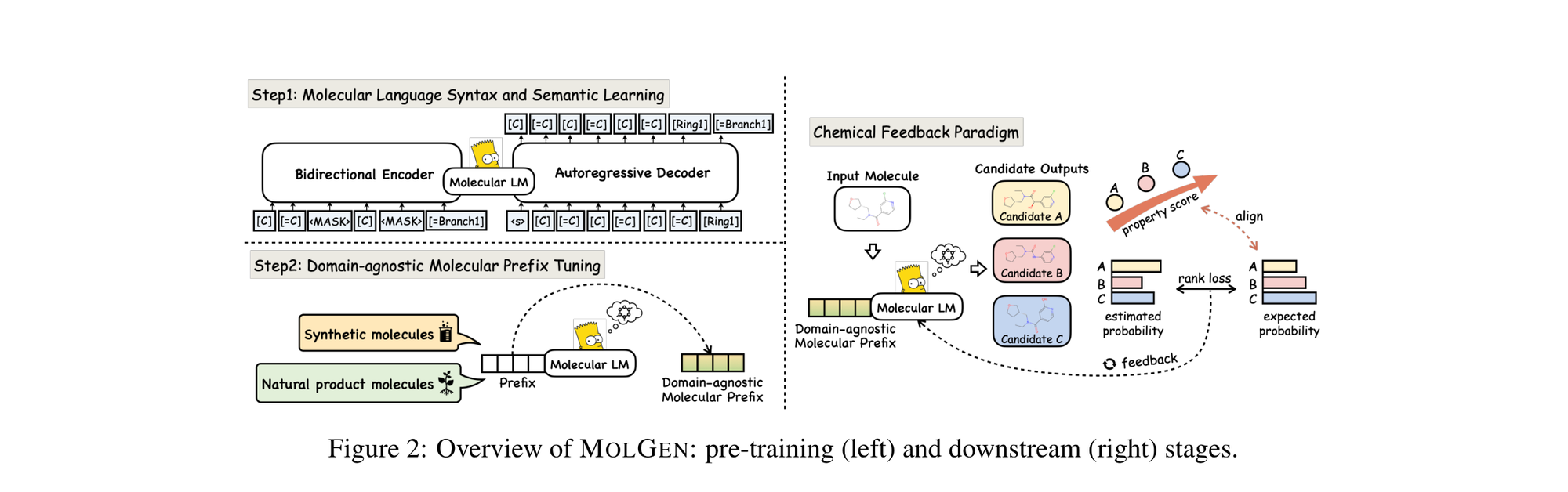
MolGen: Molecular Generation with Chemical Feedback
MolGen pre-trains on 100M+ SELFIES molecules, introduces domain-agnostic prefix tuning for cross-domain transfer, and applies a chemical feedback paradigm to reduce molecular hallucinations.
MolGen pre-trains on 100M+ SELFIES molecules, introduces domain-agnostic prefix tuning for cross-domain transfer, and applies a chemical feedback paradigm to reduce molecular hallucinations.
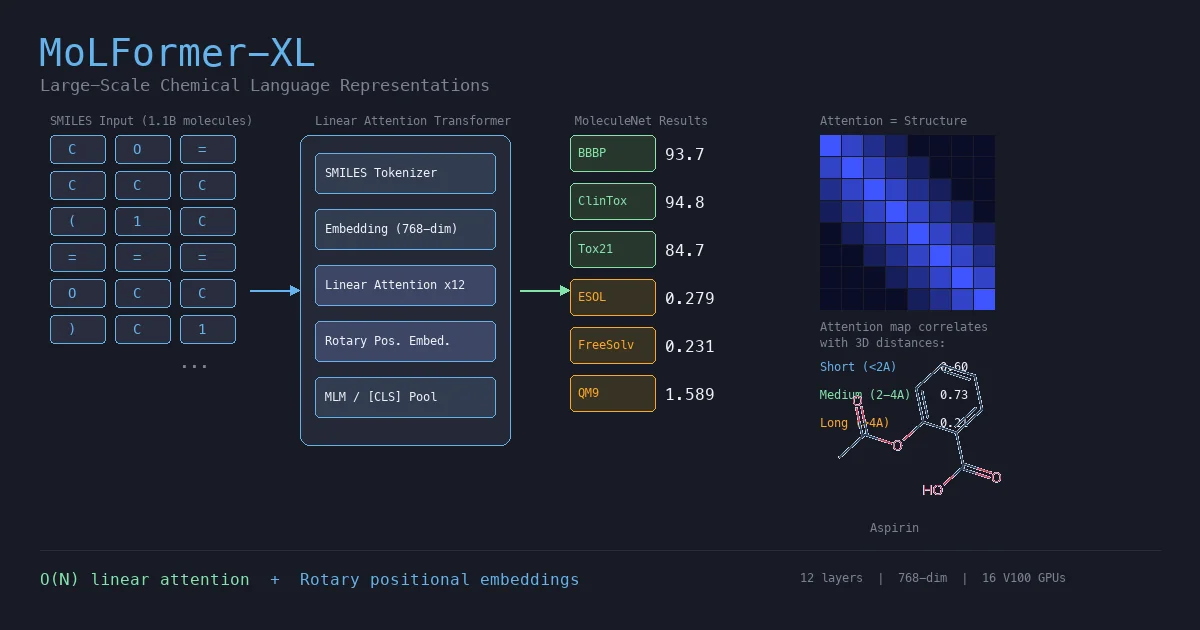
MoLFormer is a transformer encoder with linear attention and rotary positional embeddings, pretrained via masked language modeling on 1.1 billion molecules from PubChem and ZINC. MoLFormer-XL outperforms GNN baselines on most MoleculeNet classification and regression tasks, and attention analysis reveals that the model learns interatomic spatial relationships directly from SMILES strings.
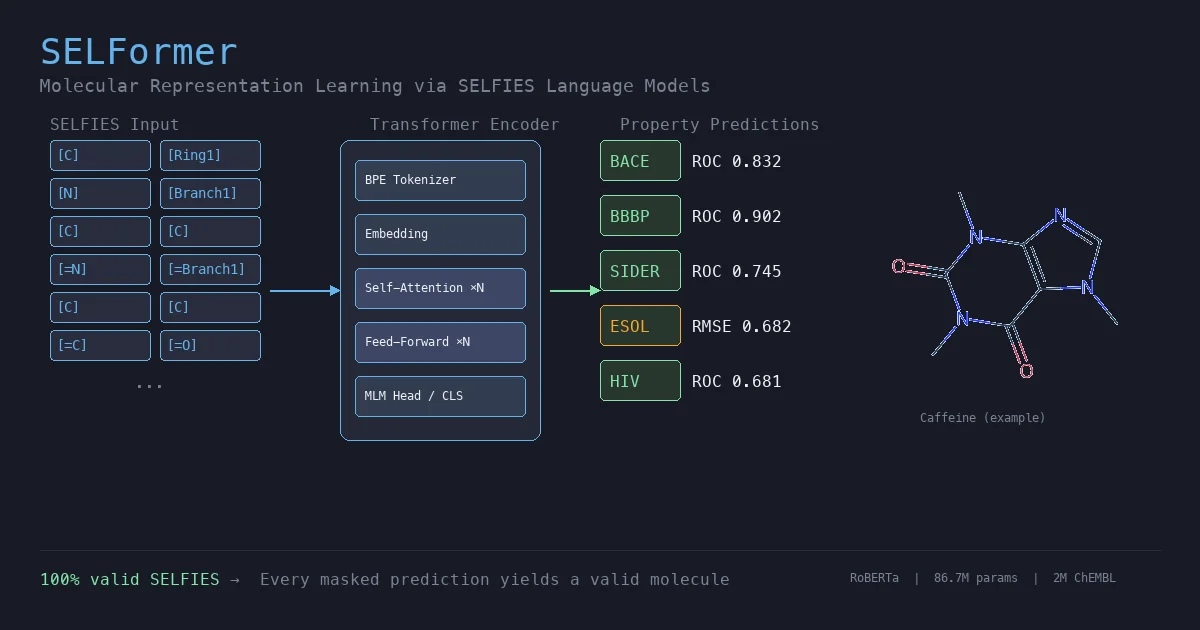
SELFormer is a transformer-based chemical language model that uses SELFIES instead of SMILES as input. Pretrained on 2M ChEMBL compounds via masked language modeling, it achieves strong classification performance on MoleculeNet tasks, outperforming ChemBERTa-2 by ~12% on average across BACE, BBBP, and HIV.

ChemBERTa-3 provides a unified, scalable infrastructure for pretraining and benchmarking chemical foundation models. It addresses reproducibility gaps in previous studies like MoLFormer through standardized scaffold splitting and open-source tooling.

This work investigates the scaling hypothesis for molecular transformers, training RoBERTa models on 77M SMILES from PubChem. It compares Masked Language Modeling (MLM) against Multi-Task Regression (MTR) pretraining, finding that MTR yields better downstream performance but is computationally heavier.
This methodological paper proposes a linear-attention transformer decoder trained on 1.1 billion molecules. It introduces pair-tuning for efficient property optimization and establishes empirical scaling laws relating inference compute to generation novelty.
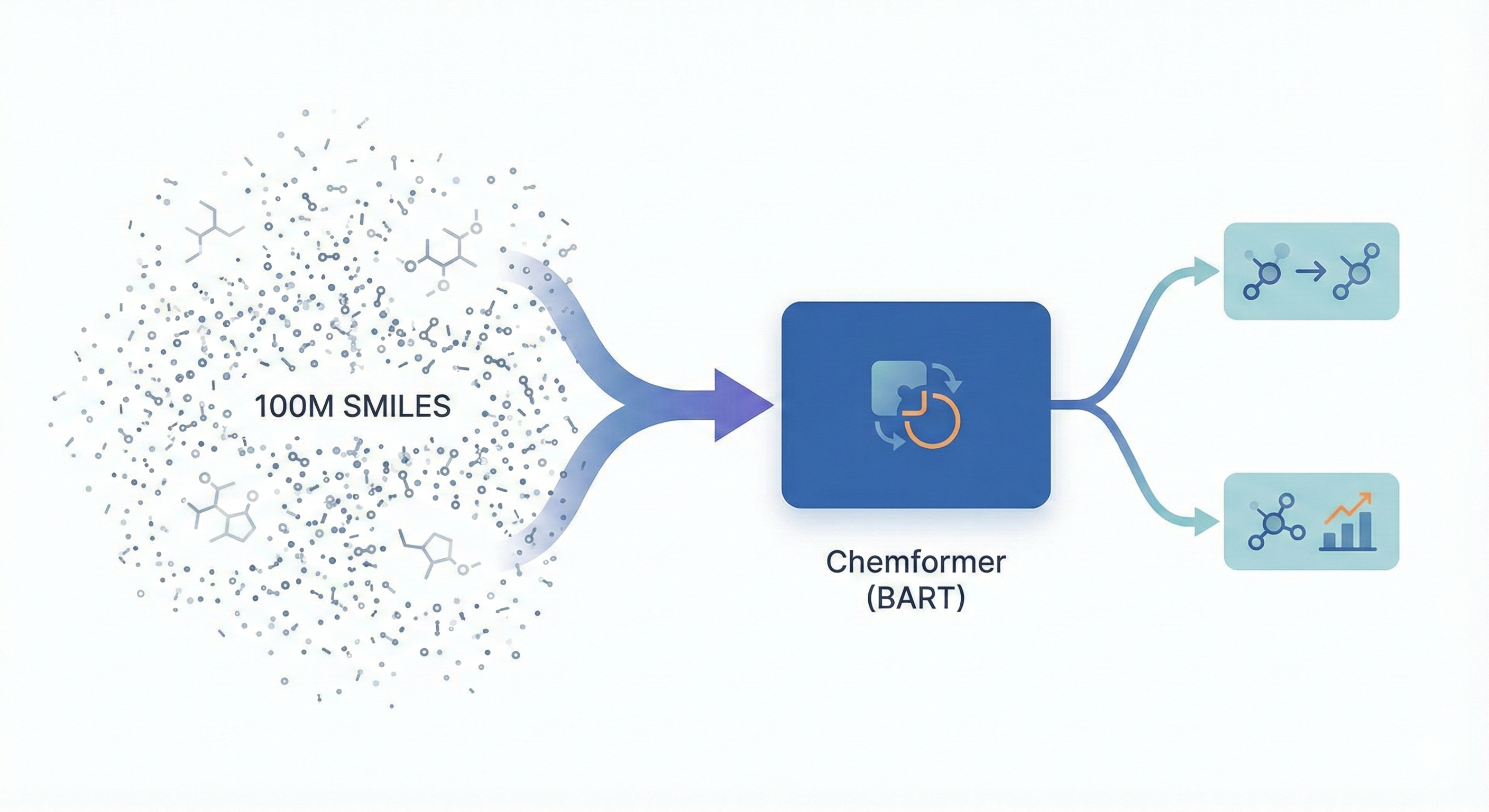
This paper introduces Chemformer, a BART-based sequence-to-sequence model pre-trained on 100M molecules using a ‘combined’ masking and augmentation task. It achieves top-1 accuracy on reaction prediction benchmarks while significantly reducing training time through transfer learning.
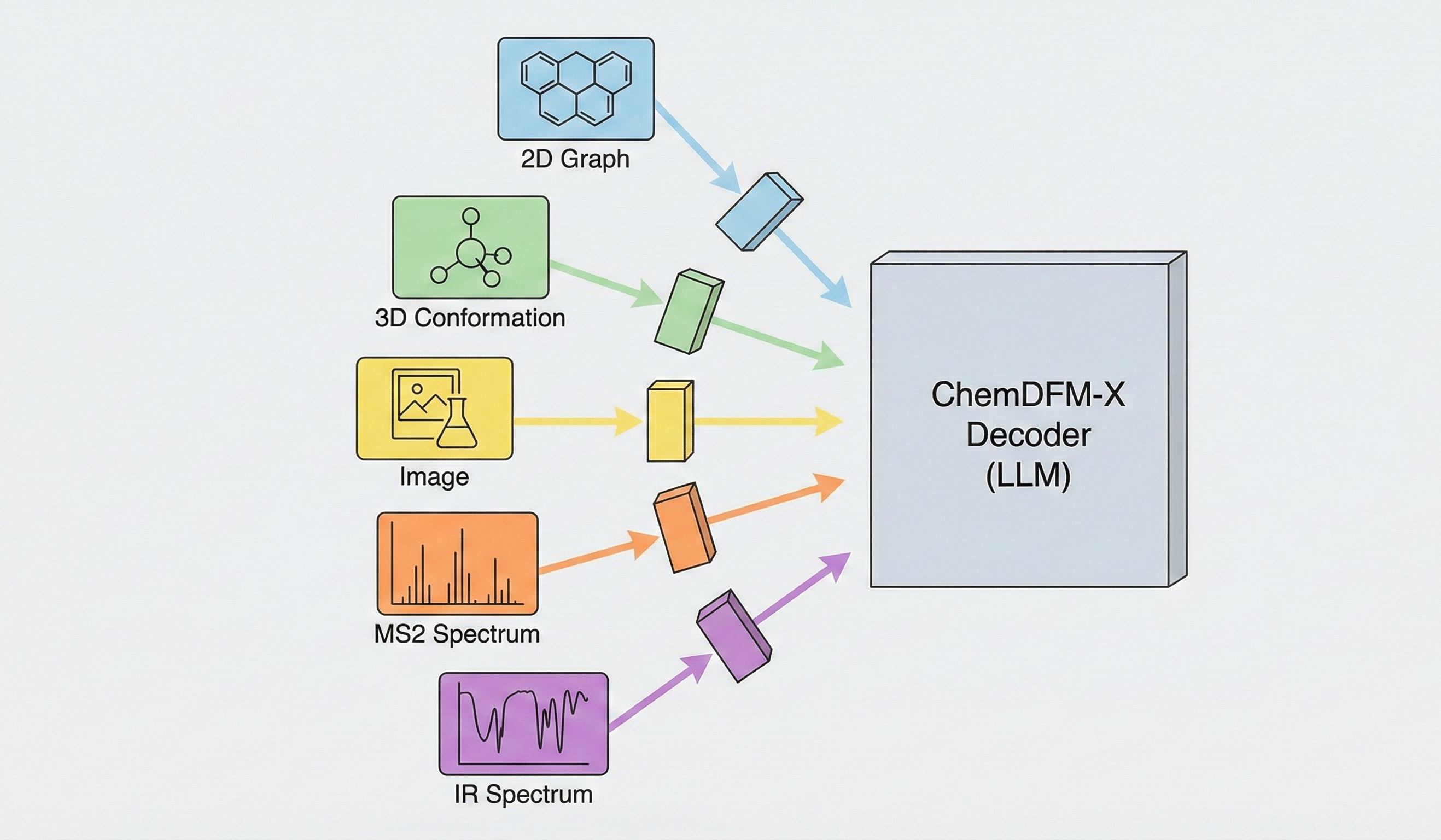
ChemDFM-X is a multimodal chemical foundation model that integrates five non-text modalities (2D graphs, 3D conformations, images, MS2 spectra, IR spectra) into a single LLM decoder. It overcomes data scarcity by generating a 7.6M instruction-tuning dataset through approximate calculations and model predictions, establishing strong baseline performance across multiple modalities.
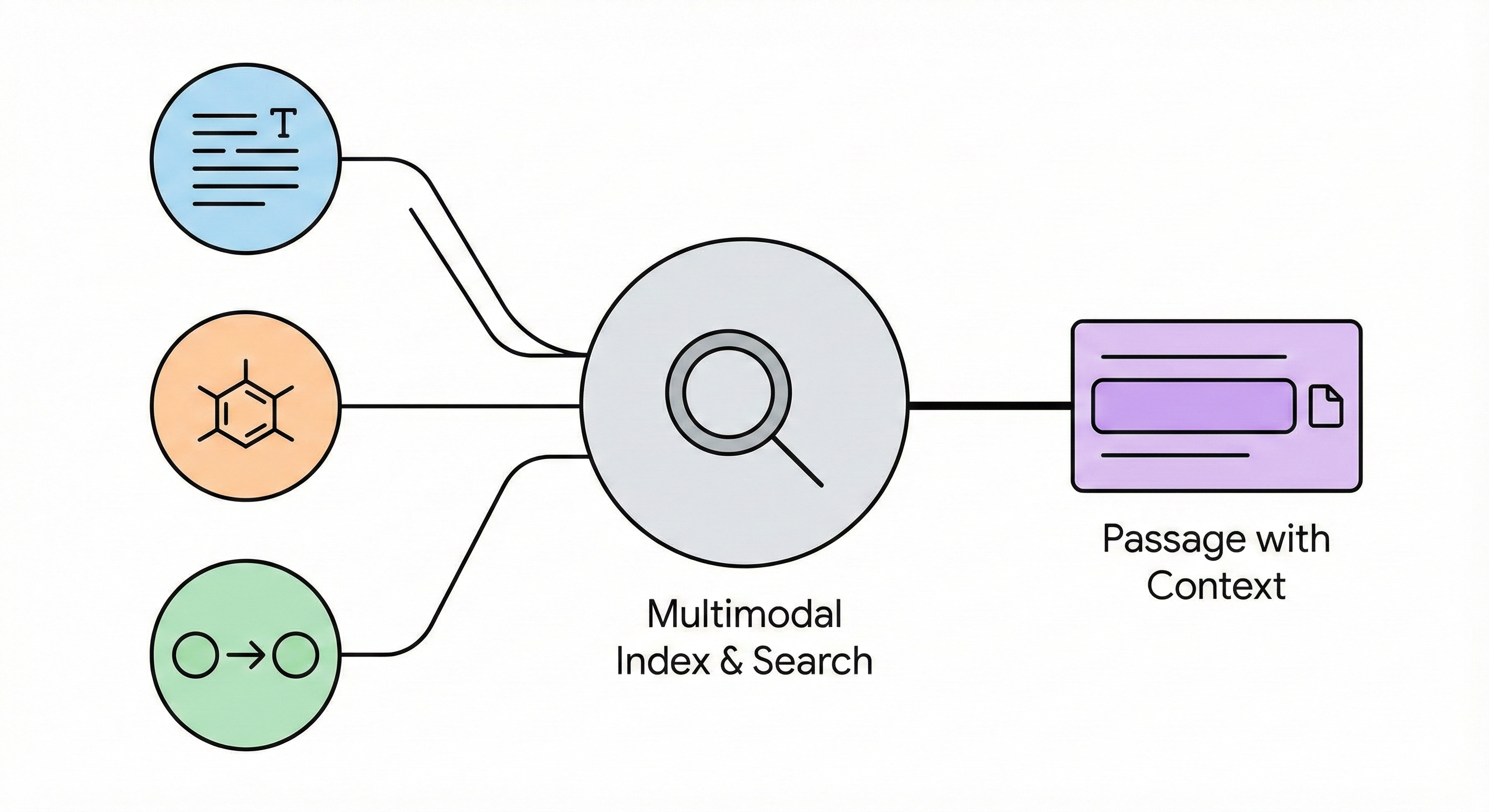
This paper presents a multimodal search system that facilitates passage-level retrieval of chemical reactions and molecular structures by linking diagrams, text, and reaction records extracted from scientific PDFs.
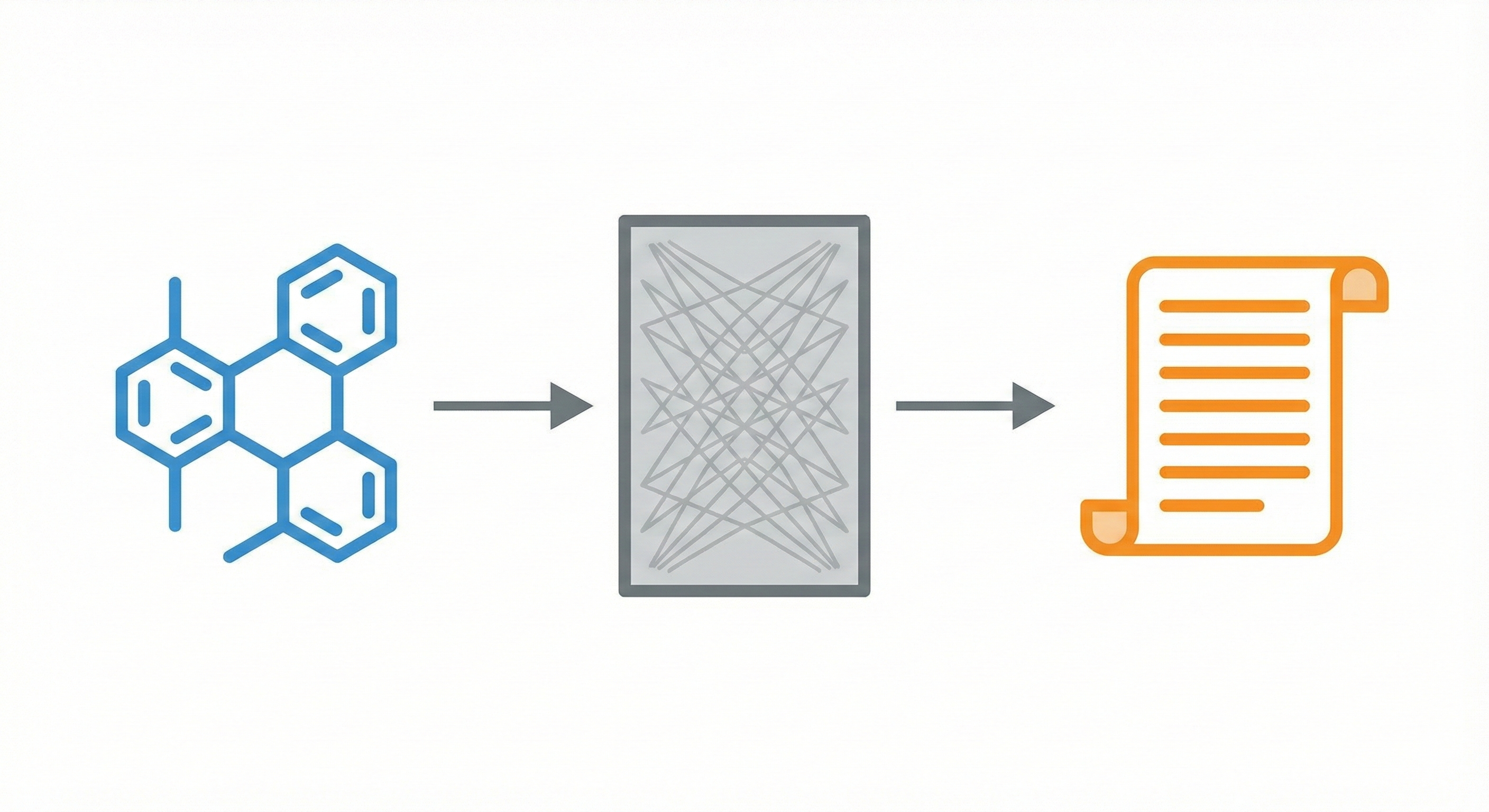
STOUT V2.0 uses Transformers trained on ~1 billion SMILES-IUPAC pairs to accurately translate chemical structures into systematic names (and vice-versa), outperforming its RNN predecessor.

STOUT (SMILES-TO-IUPAC-name translator) uses neural machine translation to convert chemical line notations to IUPAC names and vice versa, achieving ~90% BLEU score. It addresses the lack of open-source tools for algorithmic IUPAC naming.
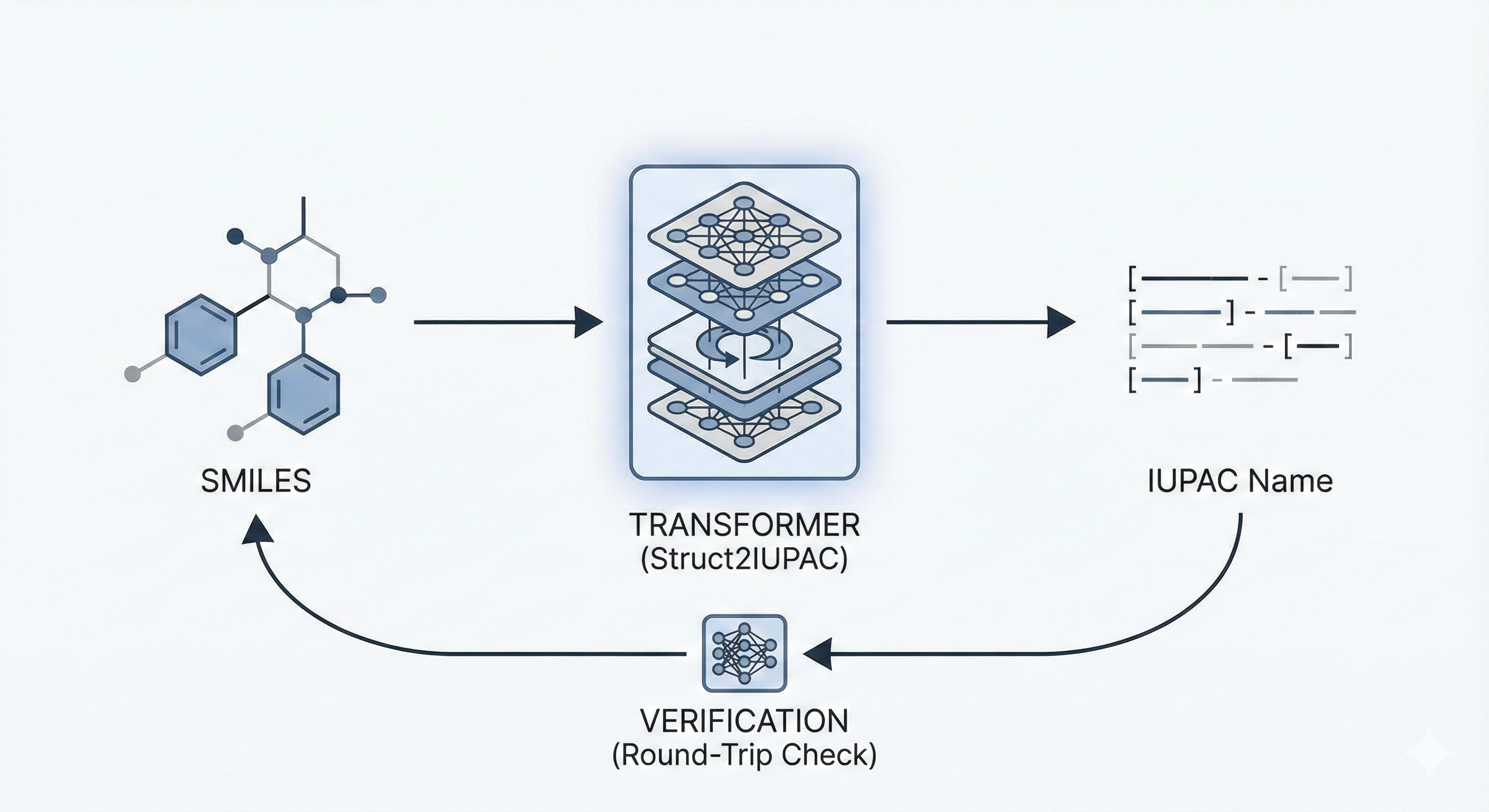
This paper proposes a Transformer-based approach (Struct2IUPAC) to convert chemical structures to IUPAC names, challenging the dominance of rule-based systems. Trained on ~47M PubChem examples, it achieves near-perfect accuracy using a round-trip verification step with OPSIN.