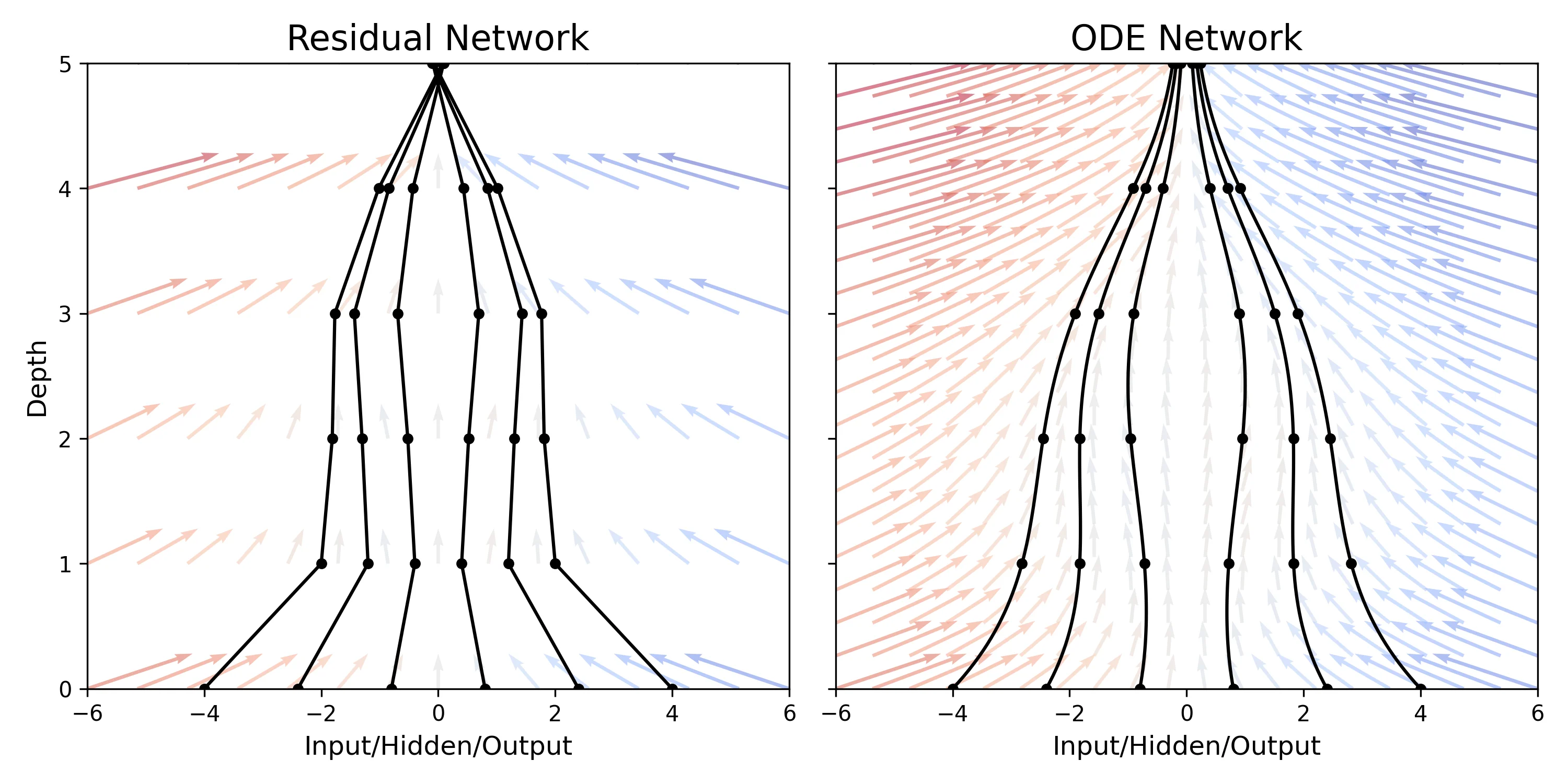
Neural ODEs: Continuous-Depth Deep Learning Models
This paper replaces discrete network layers with continuous ordinary differential equations (ODEs), allowing for adaptive computation depth and constant memory cost during training via the adjoint sensitivity method. It introduces Continuous Normalizing Flows and latent ODEs for time-series.